
| 일 | 월 | 화 | 수 | 목 | 금 | 토 |
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| 8 | 9 | 10 | 11 | 12 | 13 | 14 |
| 15 | 16 | 17 | 18 | 19 | 20 | 21 |
| 22 | 23 | 24 | 25 | 26 | 27 | 28 |
- 유전역학
- MRI
- 확산강조영상
- genetic epidemiology
- non-parametric model
- 모수적 모델
- words encoding
- tabular
- parrec
- 비모수적 모델
- paper review
- 확산텐서영상
- PYTHON
- nlp
- 파이썬
- MICCAI
- parer review
- parametric model
- Phase recognition
- monai
- precision #정밀도 #민감도 #sensitivity #특이도 #specifisity #F1 score #dice score #confusion matrix #recall #PR-AUC #ROC-AUC #PR curve #ROC curve #NPV #PPV
- nibabel
- Surgical video analysis
- 데코레이터
- deep learning #segmentation #sementic #pytorch #UNETR #transformer #UNET #3D #3D medical image
- TabNet
- 코드오류
- decorater
- nfiti
- TeCNO
- Today
- Total
KimbgAI
[medical] 유전역학-개론 4주차, 전장유전체 연관분석(GWAS) 본문
GWAS를 설명하기 위해 지난 3주차까지 개념을 익힌거라고 봐도 될만큼 이번 학기 중 어떤 한 축이되는 강의이다.
이번주는 실제 논문을 읽고 할때 도움이 되는 개념들을 많이 익힐 것이다.

계속 보여지는 슬라이드인데, Phenotype에 대한 genotype의 연관성을 보는 것이 연관 분석이고, SNP 하나만 보는 것이 아니라 매우 많은 SNP을 가지고 연관분석하는 것이 GWAS의 개념이다.
Outcome에는 Phenotype 뿐만 아니라 RNA expressure 과 같은 다양한 것들이 들어갈 수 있다.

그렇다면, Genotype에 들어갈수있는 데이터는 무엇이 있느냐? 대게 위의 4가지 정도이다.
가장 많이 사용되는 것이 GWAS Microarray이다.
SNP 마커, 약 100만개의 genetic marker로 구성된 chip array가 GWAS 연구에 쓰이고 있고, 그 이유는 가성비가 좋기 때문이다.
WGS는 그에 비해 가격이 10배 이상 차이날만큼 비싸다.
rare disease가 목적이 아니라, population 연구가 목적이기 때문에 대부분의 gene을 커버하는 GWAS가 많이 사용된다.

GWAS는 disease에 미치는 특정 gene에 대한 어떤 가설이 없이 연구를 시작하는 것이 특징이다.
candidate approach는 특정 gene이 어떤 disease에 영향이 있을 것이라고 가설을 세우고 연구를 하지만, GWAS는 그렇지 않고 common complex disease라는 가정만을 세운다.
common variant 라고 하는 것이 SNP의 정의이고, effect allele이라고 하는것은 major 하지 않은 minor allele을 말한다.
보통 common variant라고 하는 것은 MAF(minor allele frequency, 두번째로 많은 염기인 소수염기의 빈도)가 5% 이상일때로 정의하고, 1%보다 작다면 rare variant로 정의한다.
따라서 1%보다 큰 variant들은 GWAS로 찾아보겠다 하는 것.
그럼 얼마나 많은 SNP을 찾아야하는가?
보통 european 기준으로 500k~1M정도가 되면 전체 SNP의 67~89% 정도 capture할 수 있다고 한다.
'capture' 라고 하는 것은 '대략적', '추측' 의 개념이 들어있는데,
1억개의 variant들을 다 찾아내기에는 시간과 비용이 많이 들기때문에, tagSNPs 이라는 것을 정의한다. 이의 역할은 앞뒤로 그와 관련한 SNP들을 알 수 있고, 그렇기 때문에 모든 SNP을 조사할 필요없이 몇몇의 tagSNP만을 조사해도 대부분의 SNP을 알 수 있게 되는 것!
그렇다면 tagSNP을 어떻게 알아낼 수 있는가?
Linkage Disequilibrium (LD)의 개념을 활용해서 알아낼 수 있다.
예를 들면, 위 그림에서 빨간색과 노란색이 Linkage되어 있다는 것을 알 수 있으면, 빨간색을 알면 노란색을 알게되고,
16개의 SNP 중 빨간색 4개만 찾아내면 12개를 커버할 수 있는 것과 같다.
파란색은 알지 못하지만, 가성비가 매우 좋아지게 됨.

위 그림이 LD의 개념이다.
두 loci가 있고, 각 allele의 frequency가 위 그림처럼 알려져 있다고 해보자.
그럼 두 allele들로 조합이 되어 나올 수 있는 haplotype의 확률은 두 allele의 frequency를 곱한 값과 같다.
이처럼 계산된 대로 관찰이 되면 Equilibrium 상태이다. (자연적으로 기대되는 상태)
하지만 실제로 봤을때 그렇지 않다면, Disequilibrium이다.
이럴 때는 두 allele은 강하게 Linkage 되어 있다는 것을 의미한다.

LD는 r^2나 D프라임으로 측정하는데, 일반적으로 상관관계라고 생각해도 무방하고 0.8 이상인 경우 high LD라고 한다.
haplotype이 얼마나 복잡해졌냐, LD가 적게 일어났느냐 하는 것은 세대교체가 얼마나 많이 이루어졌느냐에 따라 다르다.
따라서 인류의 기원이라고 할 수 있는 아프리카계 인종들은 유러피안보다 2배정도 더 복잡하다고 한다.

위 그림을 보면, 4명의 사람의 haplotype 중 서로 다른 varient들이 6개가 존재하고, 그 비율도 모두 다르다.
6개의 서로 다른 SNP에서 나올 수 있는 haplotype은 2의 6승으로 64개 이지만, 실제로는 그렇지 않게 관찰이 된다.
그렇다는 것은 SNP끼리 서로 linkage 되어 있다는 것을 알 수 있고, 그렇다면 모든 SNP을 다 조사할 필요는 없다는 것이다.

위 그림을 보면, SNP1~4는 서로 매우 퍼펙트하게 Linkage 되었다.
SNP1에서 A가 나오면 그 뒤부터는 CGA가 무조건 나오고, 반대로 SNP1에서 G가 관찰되면 이어서 TCG가 관찰되는 것이다. 이를 묶어서 하나의 block 개념으로 본다.

이런 식으로 LD block을 살펴서 tagSNPs을 추출할 수 있다.
tagSNP은 얼마나 엄격하게 정하는지에 따라 tagSNP을 많이 뽑을지 적게 뽑을지 정하게 되고,
위 그림에서 삼각형 형태가 끝나는 부분이 block을 구분한다고 볼 수 있다.

MAF 1% 이상의 4백만개의 SNP을 조사한 결과, 유퍼피안은 30만개이상의 tagSNP이 필요한 반면, 아프리카계는 60만개 이상이 필요했다.

위 그림은 인종에 따른 LD block 수의 차이를 보여준다. r^2 를 기준으로 0.5 이상일때 LD가 보인다 라고 했을때,
아프리카계는 60만개 이상의 tagSNP 나왔지만, 아시아쪽은 27만개 정도의 tagSNP이 나온다.

GWAS의 특징은 유의미하게 나온 SNP이 바로 causal allele이 아니라 indirect association으로 그 SNP 근처의 있는 것이 causal variant이다. 찾은 SNP은 LD로 인한 그저 tagSNP이기 때문이다.

Genotyping은 마커를 사용하고, 500k인 경우 50만개의 비즈가 심어져 있고, 한 비즈가 한 SNP marker이다.
한 SNP marker면 두가지 allele 정보를 가진다.
한 allele에는 초록색 형광 물질을 두고, 다른 allele에는 빨간색 형광 물질을 둔다.
그래서 같은 allele(homozygote)이면 초록색, alternative allele(another homozygote)면 빨간색, heterozygote이면 노란색으로 나타난다.

전체적인 flow는 위 그림과 같다.
Imputation 개념은 뒤에 더 설명하겠지만, 내 연구의 sample case가 모자라서 다른 사람의 연구결과를 활용하여 case를 조금 더 늘리는 방법인데, 서로 같은 chip array가 아니기 때문에 이를 보정하는 작업이 필요하다.
이 작업을 imputation이라고 한다.

위 논문은 최초의 GWAS 컨소시움이고 여기서 WTCCC라는 개념을 통해 QC 등과 같은 분석 파이프라인을 이 연구에서 정석으로 정하였기 때문에, starting point paper로 아주 좋다고 한다.

GWAS는 앞에서 DNA를 추출하고, genotyping을 하고 등의 단계를 거치고 난 후, 실제로는 그저 Statistical anaysis 단계이다. 많은 저널들은 이 단계가 끝나고 재현성 검증을 위해 replication과 메타분석을 요구하고 있다.
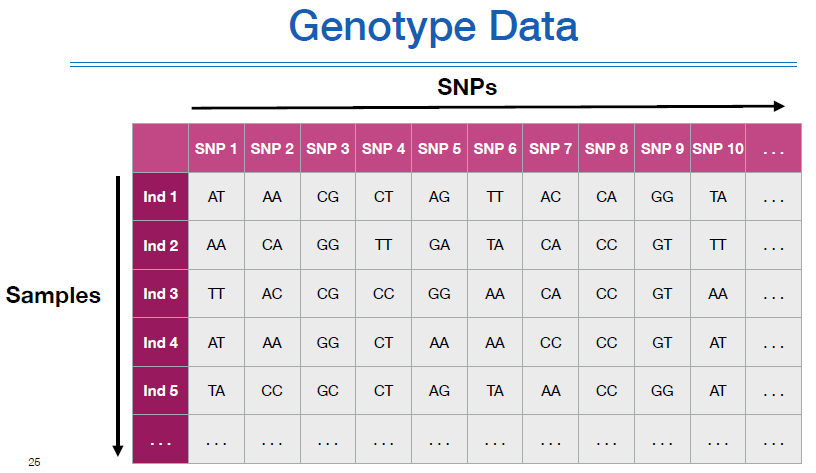
저번주차에 언급했던것처럼 genotype data를 실제로 받으면 위와 같다. (엑셀에서 안 열림, 파일이 너무 커서)
보는것처럼 diplotype으로 되어있다.
이렇게 데이터를 받았다면 QC를 해야한다.

QC는 sample QC와 SNP QC 두가지 방향으로 진행된다.
제일 먼저 체크하는게 sample QC 중 성별체크이다.
DNA 추출을 위한 sample들의 인터뷰지와 같은 임상정보와 실제 genotype이 성별이 서로 다르다면, 이럴때는 무조건 생물학적인 정보인 genotype을 믿어야한다.
하지만 임상정보와 genotype이 불일치하기 때문에 버려야하는 샘플이다..ㅠ
genotype call rate는 genotyping 할때 얼마나 잘 뽑아졌는지가 call rate이다. 보통 95%를 call rate로 본다. 위 그림에서 00은 missing data을 의미한다.
또한, 가족들의 표본을 제외해야하는 것도 정석이다.
또 SNP QC에서는 HWE도 하고, rare한 MAF도 버린다.
어쨌든 이렇게 QC가 끝나면 아래와 같은 프로그램에 넣어 돌리면 끝이다.

PLINK가 제일 빠르고 유명하다.
또 각자만의 특성이 있어 그에 맞게 사용하는 것이 중요하다.

GWAS의 통계분석은 두가지가 있다.
케이스-컨트롤 분석은 로지스틱 회귀분석의 결과를 카이제곱검정으로 테스트한다.
정량적 분석은 선형회귀분석을 한다.
보통 additive model로 분석한다.
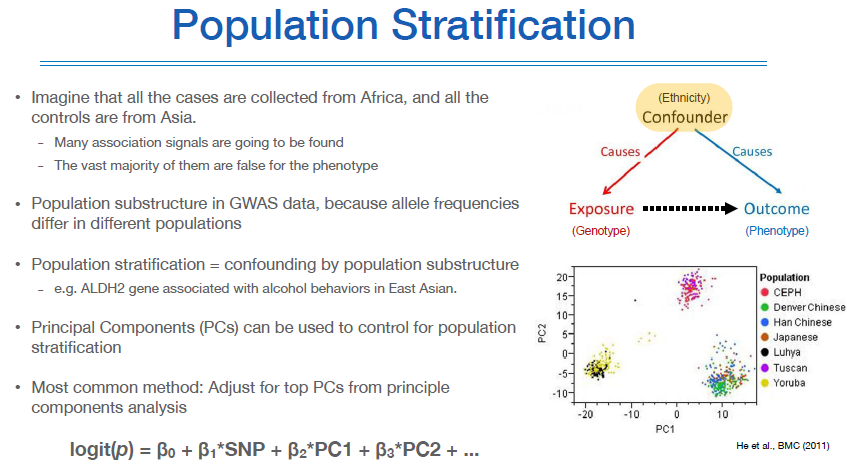
컴파운더 조정을 위해 population stratification (인종간의 구분되는 특징)을 봐야한다.
이를 위해 PCA(주성분분석)을 수행하고, 이런 주성븐은 내가 연구하는 인구특성을 몇개 대표해서 보여준다.
예를들면 500만개의 SNP을 2~3개의 SNP 정보로 대표해서 보여주는 것이다.
이렇게 하면 우측 아래 그림처럼 ploting이 된다.
이런 주성분들을 분석에 있어서 보정변수로 넣어 분석을 한다.

위 그림은 인종별로 얼마나 주성분이 분포되어 있는지를 보여주는 그림이다.
한국(초록색 점섬)은 대체로 오밀조밀하게 모여있다. 하지만 다른 나라들은 다양한 인종이 섞여있어 폭넓게 분포하기도 한다.

결과가 나오면, 대체로 엑셀이나 이런것들을 활용하여 결과를 확인하기 어렵고,
이 모든 것들을 다 논문에 실을수 없기 때문에, 중요한 것이 무엇인지 확인하고 선별하는 과정이 필요하다.
대게 위와 같은 프로세스를 수행한다.

가장 먼저 보는 것이 QQ plot이다.
QQ plot은 x 축은 랜덤한 데이터를 가지고 pvalue를 구했을때의 pvalue의 기대값 분포이고 y축은 관찰값을 sorting한 것이다.
따라서 (a)와 같은 그림이 나타나면, 내 데이터는 매우 낮은 연관성을 보이는 것이다. 말그대로 랜덤한 결과..
이럴때는 샘플수가 적어서 그럴 가능성이 높다.
(b) 와 같이 축에 붙어서 가지않고 처음부터 뜬다면, population stratification이 있다는 것이다.
이럴 때는 sample QC를 다시 해야한다.
(d) 와 같은것이 이상적이다.
근데 구체적으로 어떤 SNP인지 모르기 때문에 아래와 같은 것을 수행한다.

맨하탄 plot은 미국 맨하탄의 마천루 모습과 비슷하여 지어진 이름인다.
점 하나가 SNP 하나를 의미한다.
6번 염색체의 앞쪽 부분의 SNP가 무언가 유의미하게 나온것을 볼 수 있다.
pvalue의 기준인 빨간색선은 5x10e-8이다. 왜 0.05가 아니고 저렇게 엄격한 기준을 적용하냐면, 분석하는 대상이 보통 100만개 정도로 너무 많기 때문이다. 자세한건 뒤에.
저렇게 우뚝 솓은것이 하나의 줄이 아니고 확대하면 아래와 같이 보인다.

빨간색일수록 높은 LD를 보여준다.
tagSNP은 블럭에서 하나만 뽑는 것이 아니기 때문에 저렇게 강한 LD를 보이는 SNP가 여러개 있다.
x축은 SNP의 더 자세한 위치를 나타낸다.
결과를 보면 왼쪽 아프리카계 인종은 유의한 것이 많지 않은데, 중간의 유러피안은 유의미하게 나타나는 것을 볼 수 있다.

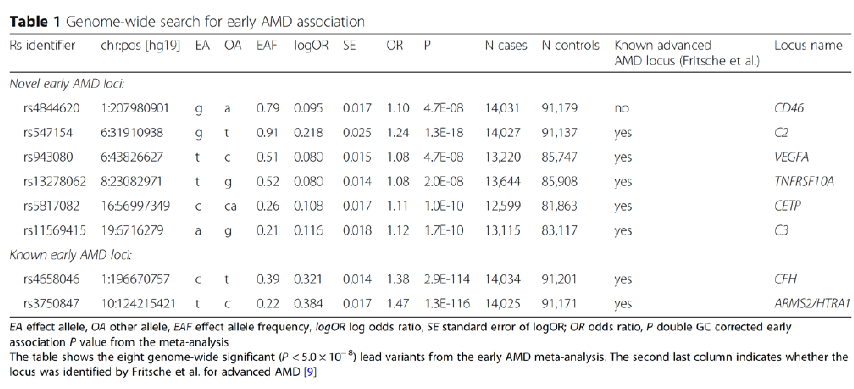
결과 테이블이다.
보통 EA(effect allele)은 minor allele을 말하는데, 여기서 EAF(effect allele frequency)를 보면 major allele을 말하는 것 같다.
case와 control이 있는 것으로 보아 case-control 분석이며, 정량분석이면 베타값이 기재된다.
위 표의 좌측처럼 SNP은 rs머시기로 명명되는데, 해당 SNP은 아래 dbSNP에서 해당 SNP과 관련된 정보를 찾을 수 있다.

GWAS에서는 일반적으로 통계에서 사용하는 pvalue인 0.05를 사용하지 않는다.
이유는 아래와 같다.

독립분석을 보통 100만번 하기 때문이다. 0.05라고 하는 pvalue는 통상 100번의 같은 연구를 했을때 95번정도는 같은 결과가 나올것이다 하는 의미인데, 100만번의 분석은 FDR가 너무너무 많아지기 때문임.
말 그대로 무작위 SNP 100만개를 돌렸을때 0.05로 하면 그중 5만개는 유의미하게 나올 수도 있기 때문이다.
그래서 이를 보정하기 위해 분석하는 횟수만큼 나눠준다.
샘플사이즈가 작으면 이런 엄격한 유의수준을 넘어서는 애들이 별로 없기 때문에 샘플사이즈가 그래서 중요하다.

유의미하게 나오지 않았더라도, 비슷한 유의수준을 보이면 보고를 할 수 있는데,
그러기 위해서는 False positive의 위험을 방지하고, 재현성 검증을 위해 replication study가 필요하다.
이때는 모든 SNP을 다 하는 것이 아니라, 새로운 sample를 모집해서 (대체로 1000 sample?) 다시 독립분석을 진행하는 것이다.
몇개의 후보 SNP만 하기때문에 적은 비용으로 할 수 있다는 장점이 있다.

위 표는 replication study를 두번한 결과이다.
하지만 이제는 리뷰어들이 replication study보다 meta analysis를 요구한다고 한다.
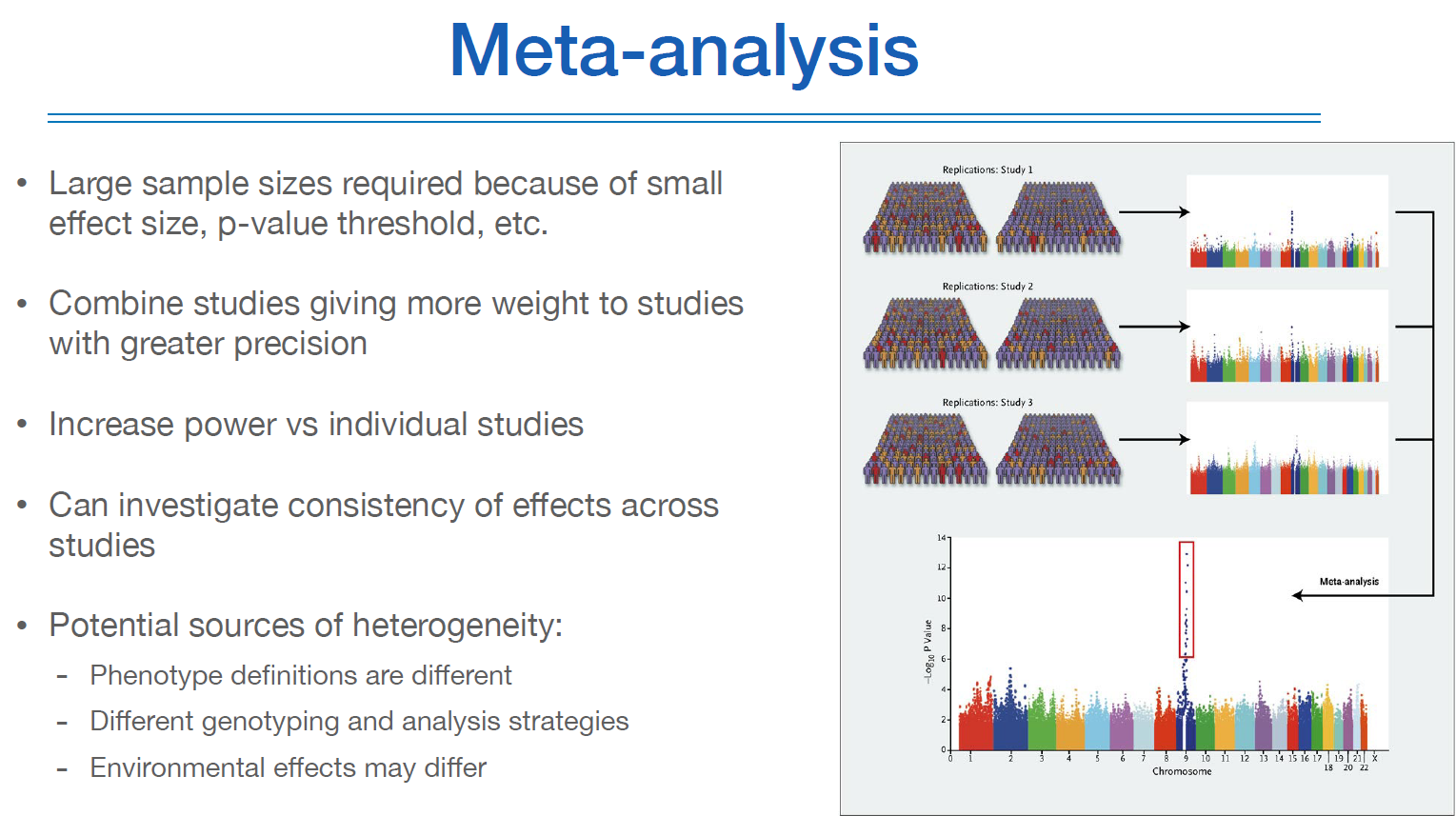
메타분석은 위 그림에서 우측 그림처럼 replication 연구를 여러번 했는데도 유의미하게 나오지 않았지만, 그 모든 연구의 표본을 합쳐서 보니 유의미하게 나왔다는 것이다.
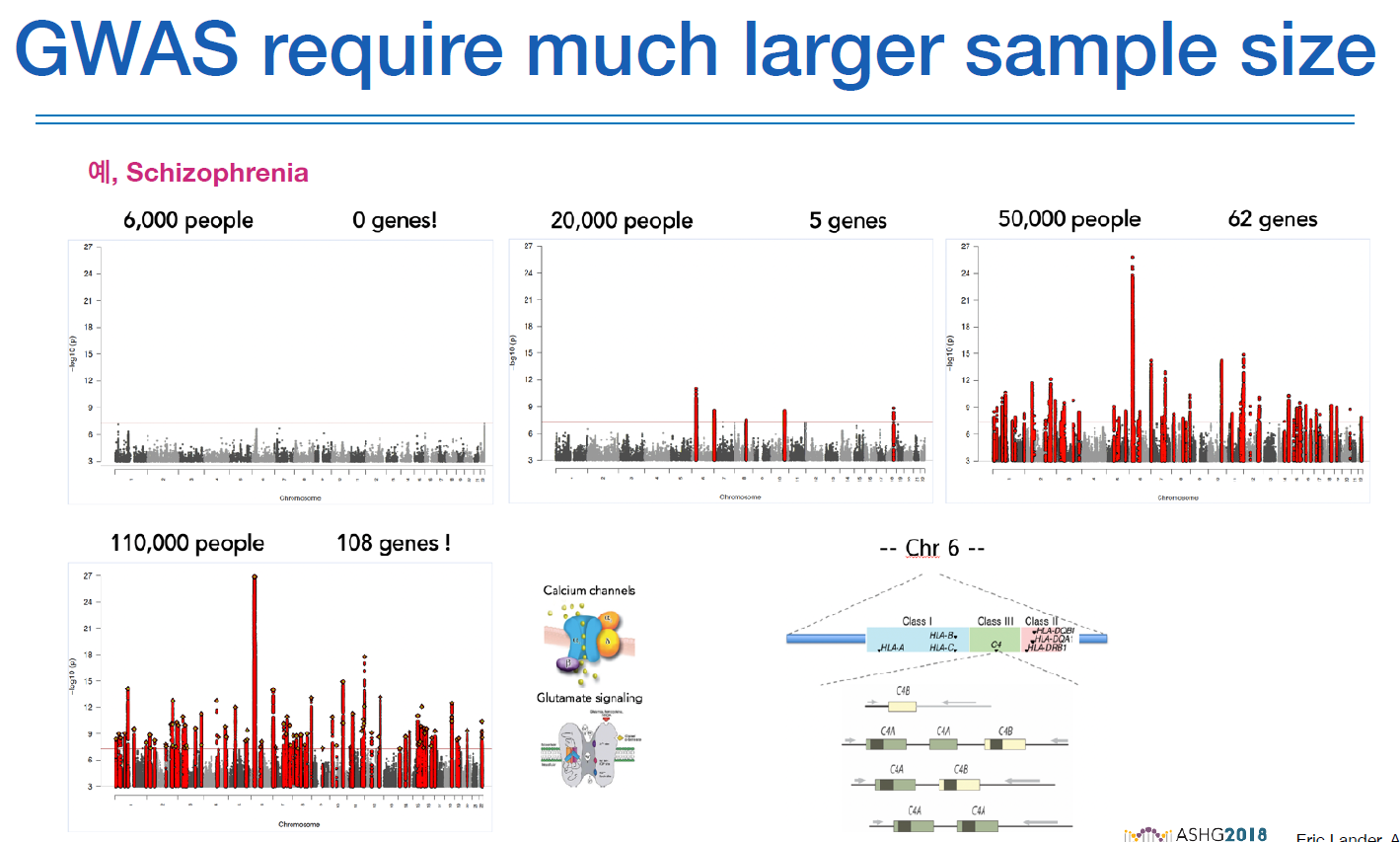
11만명의 케이스에서 많은 SNP들이 true로 나왔고, 이게 전부다 true인가? 하면 대체로 그렇다. 그렇기 때문에 complex disease인것.

근데 다른 연구와 합치려고 하니 연구마다 제네틱마커가 제각각이라 어려운 점이 있다.
그래서 imputation study를 하면 교집합이 조금더 넓어질수있다.

내가 가지고 있는 haplotype과 레퍼런스를 비교하고 linkage 개념을 이용하여, 같은 allele이 나타나면 빈 자리를 레퍼런스를 통해 매꾸는 개념이다.
그래서 이걸 얼마나 엄격하게 하느냐, imputation rate를 정하느냐에 따라 imputation quality를 정한다. (0~1)

메타분석은 위와 같이 이루어지고 보고를 한다.
Source에서의 행 하나하나가 한 코호트들이다. 각각 array 칩이 다른걸 볼수있고, 같은 370k 칩이라도 같은 회사의 것이 아닐 수 있다.
그래서 보면 600K 칩이 imputation을 하니까 8M칩으로 바뀐걸 볼 수 있다.
두번째 테이블을 보면, 앞서 본인이 했던 연구에서 밝히고 싶은 SNP들을 imputation한 연구들과 비교했을때, direction을 보면 +는 positive, -는 negative을 의미하고, 만약 해당 SNP가 imputation이 안되었으면 빈칸이나 물음표로 기재된다.
다른 연구들의 결과와 같이 +가 많이 나오면 좋은 것!

많은 GWAS 연구에서 밝혀진 결과들 중 유의미한 것들은 GWAS catalog에 다 올라가있다.
그래서 질환을 검색하면 질환과 관련된 SNP들이 나오고, SNP을 검색하면 그 SNP과 관련한 질환들이 나온다.

앞서 설명한것처럼, 찾은 SNP가 causal variant가 아님에 유념해야한다.
추가적인 방법을 이용해서 그 주변을 찾아야한다.
아무리 회사에서 functional tagSNP array를 만들었더라도, 대부분의 tagSNP은 non-protein-coding region 이다.
전체 30억 염기서열 중에 오직 2%정도만이 coding region이기 때문이다.
coding region 이라는 것은 실제로 action을 위한 단백질을 만드는 영역이라는 것이다.
그럼에도 불구하고 SNP들이 유의미하게 나오는 이유는 cis-regulatory elements 이기 때문이다.
실제 causal virant와 멀리 떨어져있더라도 그 변이의 function을 어느정도 조절할 수 있다.

아무튼 GWAS 연구는 여기서 끝이고, 여기서 나온 결과들을 어떻게 해석할 것인가는 다음 문제이다.
이를 위해 위와 같은 DB를 사용해서 의미를 찾는다.
근데 일일이 하나씩 찾아야하는 문제가 있기 때문에 요즘에는 아래와 같은 FUMA라는 툴을 사용하여, 나의 GWAS결과만 업로드하면 자동으로 annotation을 해준다고 한다.


위와 같은 gene-based 분석도 있다. SNP 만을 가지고는 하나하나 설명하기 어려워서(유의미하지 않기때문에) 이를 gene 단위로 묶어서 설명하는 방법이다. (gene은 우리몸에 2만개정도밖에 없기 때문에)
그럼 분석횟수가 2만번밖에 안되기 때문에, condense한 분석이다.
gene도 다시 pathway로 묶을 수가 있는데, gene 5개가 묶여서 어떤 pathway를 이루는데 이것도 다 DB상에서 정리가 되어있다. 어떤어떤 유전자들이 만나면 어떤 pathway를 보여준다던지 하는..
여하간 이런건 2학기에 실습을 할 예정!


GWAS로 많은 연구 성과를 얻었는데, 예를들면 알츠하이머 disease는 뉴런과 관련이 있을 줄 알고 그쪽에 포커싱을 했는데 알고보니 microglia 에 중요한 역할이 있다는 것을 밝혀냈다.

GWAS의 한계는 무엇일까?
먼저, sample size의 크기에 의존하다보니 유의미한 것을 찾기 힘들고, 대안으로 polygenic risk score을 활용하기도 한다.
그리고 샘플사이즈 문제로 인해 재현성이 어렵다는 점.
또한 SNP 하나하나는 보통의 효과만을 가진다. 대안으로 gene, pathway based을 활용하기도 함.
non-coding region이 많지만, non-coding region도 다양한 역할을 한다는 것을 ENCODE DB 등을 활용해 설명할 수 있다.


큰 GWAS 연구는 위와 같이 다양하게 있다.

국내에는 위와같은 것이 있다.



Post-GWAS 전까지 살펴보면 지금까지 배운 GWAS 연구이다.
몇가지 질문을 했는데,
Q1. genotype data에서 AT 와 TA를 같은 것으로 보는지?
A1. 그렇다. diplotype에서 어떤 allele이 엄마로부터 왔는지, 아빠로부터 왔는지 모르기 때문에 같은 것으로 본다.
Q2. 실제로 SNP끼리 연광성이 없기때문에 독립을 가정하고 독립분석을 하는 것인가?
A2. 그렇지 않다. 실제로 SNP끼리 연관성이 어느정도 있지만, 없다고 가정을 하고 연관분석을 시행한다.
그렇기 때문에 유의수준을 굉장히 엄격하게 보는것이다.
끝!
'medical' 카테고리의 다른 글
| [medical] 유전역학-개론 6주차, 차세대 시퀀싱(NGS) 데이터 분석 (1) | 2023.04.12 |
|---|---|
| [medical] 유전역학-개론 5주차, 유전체 시퀀싱 기술 (2) | 2023.04.05 |
| [medical] 유전역학-개론 3주차, 연구설계방법 (0) | 2023.03.22 |
| [medical] 유전역학-개론 2주차, 기초유전학 (0) | 2023.03.21 |
| [medical] 유전역학-개론 1주차, 유전역학 개요 (0) | 2023.03.14 |



