
| 일 | 월 | 화 | 수 | 목 | 금 | 토 |
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| 8 | 9 | 10 | 11 | 12 | 13 | 14 |
| 15 | 16 | 17 | 18 | 19 | 20 | 21 |
| 22 | 23 | 24 | 25 | 26 | 27 | 28 |
| 29 | 30 |
- decorater
- paper review
- TabNet
- parametric model
- 데코레이터
- nibabel
- words encoding
- genetic epidemiology
- 파이썬
- tabular
- MRI
- non-parametric model
- Phase recognition
- parer review
- Surgical video analysis
- nlp
- TeCNO
- deep learning #segmentation #sementic #pytorch #UNETR #transformer #UNET #3D #3D medical image
- 모수적 모델
- PYTHON
- monai
- 유전역학
- 확산텐서영상
- precision #정밀도 #민감도 #sensitivity #특이도 #specifisity #F1 score #dice score #confusion matrix #recall #PR-AUC #ROC-AUC #PR curve #ROC curve #NPV #PPV
- MICCAI
- 코드오류
- 확산강조영상
- parrec
- 비모수적 모델
- nfiti
- Today
- Total
KimbgAI
[medical] 유전역학-개론 5주차, 유전체 시퀀싱 기술 본문
5주차로써 전반적인 유전역학개론의 수업 절반이 지났다. 벌써? ㅎㄷㄷ
유전체 시퀀싱 기술은 말 그대로 ATGC라는 염기서열을 읽는 기술이다.
시간에 따라 기술발전이 엄청나게 이루워졌고, 유전역학의 발달은 기술의 발달과 늘 맥락을 같이한다.

저번 시간에 배운 genotyping은 genetic marker를 쓰기때문에 염기서열의 연속성이 없다.
하지만 시퀀싱은 하나하나 염기서열의 연속성을 가지고 읽을 수 있다.
하지만 현재 기술로는 통째로 30억개의 염기서열을 다 읽을 수가 없기 때문에 조각조각 정보만 잘라서 시퀀싱을 할 수 있다. 이걸 read 라고 한다.
보통 100bp에서 20kp(100개에서 2만개)를 읽고, 이를 다시 통합하여 하나의 DNA 정보 구성한다.
순차적 시퀀싱을 하는게 기본인데 high-throughput 시퀀싱은 이를 병행하여 할 수 있다. 이게 나중에 배울 NGS(차세대 시퀀싱)이다.

시퀀싱의 역사를 살펴보면, 1953년 DNA 구조를 알아냈고, 15년 뒤 최초로 시퀀싱을 한 시기는 1968년 12bp를 시퀀싱한 것이다.
시퀀싱은 protein부터 RNA 그리고 마지막에 DNA 시퀀싱에 성공을 했다.
이후 생어라는 사람이 1977년에 생어 시퀀싱을 만들어냈고, 이 기술을 이용하여 지놈 프로젝트가 시작되었다.

시퀀싱 기술은 크게 생어시퀀싱, NGS, TGS 로 3개의 세대로 나눠볼수 있다.
앞서 말한 것 처럼, 어떤 세대의 시퀀싱이든 30억개의 염기서열을 통으로 읽을 수는 없고 DNA fragmentation을 하여 잘라서 읽는 과정이 필요하다.
세대를 구분하는 기준은 바로 amplication(증폭)하는 과정에 있다.
염기서열을 읽을때 보다 정확하게 읽기 위해서 염기서열을 증폭시켜야 한다. 각각의 신호는 약하기 때문이다.
1세대와 2세대의 차이는 amplication을 얼마나 많이, 병행하여 수행했는가에 따라 나누고,
3세대는 amplication 과정이 필요없다는게 특징이다.
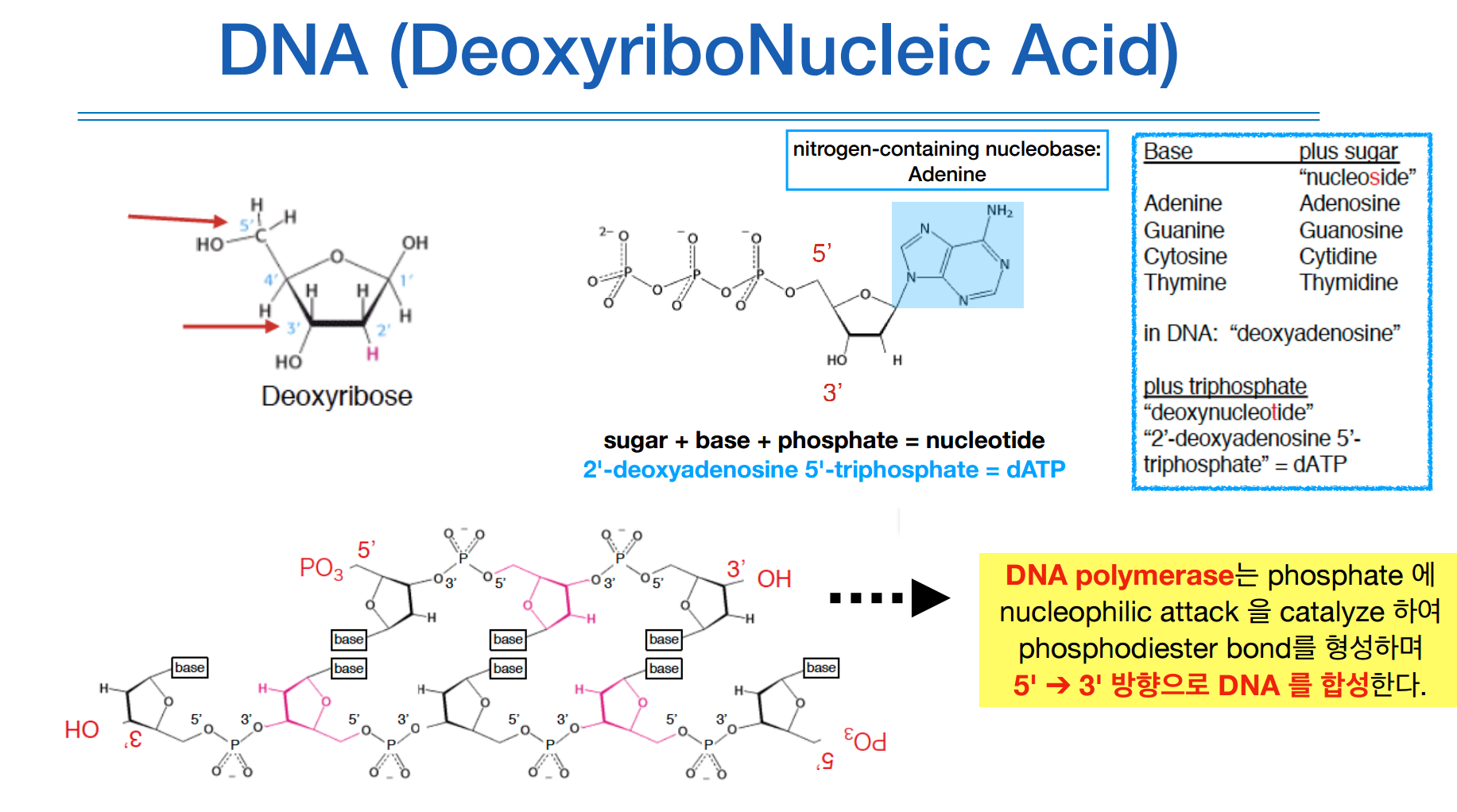
이 부분은 사실 정확히는 이해가 안간다.
맥락적인 부분을 보자면 1주차 기초유전학에서 배웠던 부분에서, DNA 합성은 5`에서 3` 방향으로 이루어 지는데 이때 산소 원자 하나가 빠진 Deoxyribose 상태에서 어떤 base가 붙느냐에 따라 ATCG가 결정된다.
이들의 이름은 위 그림에서 파란색 글씨처럼 길지만 줄여서 보면 dATP, dTTP, dCTP, dGTP가 본래 이름이고 이걸 더 줄여서 편하게 ATCG로 부르는 것이다. 이를 통칭하는 말은 dNTP이다.
DNA의 합성을 위해서는 DNA polymerase(중합효소)라는게 필요하다. 얘는 DNA의 복제를 돕는 효소이다.
생어 시퀀싱의 원리는 이러한 DNA의 합성원리를 이용한다.
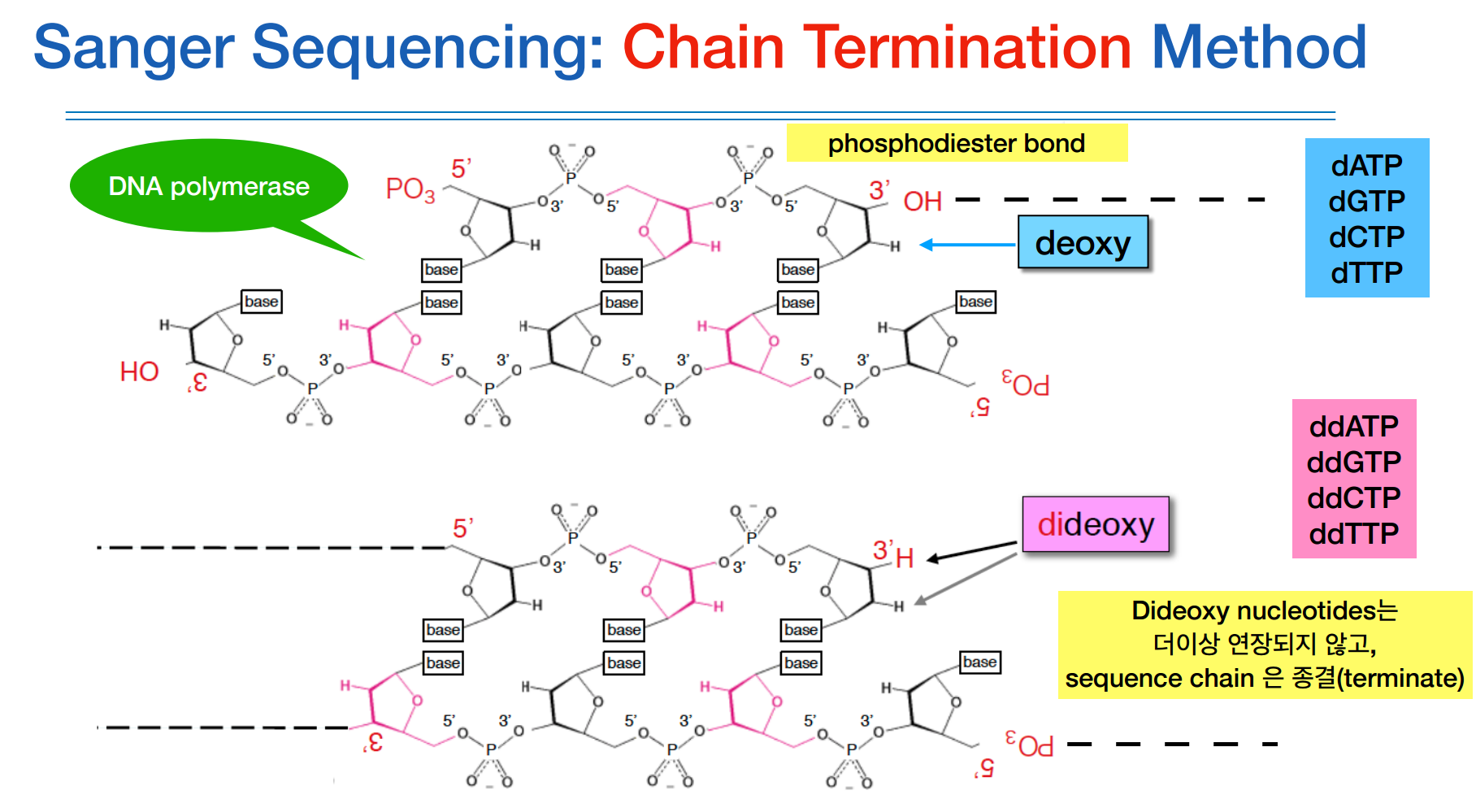
보다 구체적으로 설명하자면, DNA를 인위적으로 합성시킬때 dNTP와 DNA polymerase를 넣어주어야한다.
이때 의도적으로 합성을 방해하기 위한 ddNTP들을 넣어준다.
원래는 OH에 dNTP들이 붙어야하는데 ddNTP는 산소원자가 빠진 H만 두개가 있다. 그리하여 이 ddNTP가 붙게되면 더이상 합성이 이루어지지 못하고 중단된다.
이 원리가 chain termination 방법이다.
합성이 종결되면 ddNTP들에 각각의 형광물질을 붙혀 신호를 확인하여 어떤 서열로 종결이 되었는지 알 수 있다.

생어 메소드는 위처럼 크게 6가지 스텝으로 진행된다.
먼저 1) DNA를 자르고 2) DNA를 증폭시킨다.
DNA 증폭은 생어 방법에서는 박테리아를 통해 증폭을 시키는 방법을 사용하고, 이를 in vivo cloning이라고 한다.
3) Denaturation은 DNA는 상보서열이기 때문에 하나의 lane만 관찰하기 위해 ssDNA로 만드는 것이다.
4) Primer는 DNA의 합성이 출발점을 말한다. 짧은 올리고뉴클로타이드를 만들어서 이를 스타터로 사용하고, 앞서 말한 dNTP와 polymerase 그리고 ddNTP를 다 집어넣어 합성을 시작한다.
5) 합성을 시작하면 중간중간 ddNTP가 붙어 합성 종결이 일어나고
6) electrophoresis(전기영동)을 통해 확인을 할 수 있다.

생어 시퀀싱을 하기 위한 준비물들은 위와 같다.
예전에는 방사성 동위원소로 각 ddNTP에 라벨을 했지만 요즘에는 형광물질을 바른다.
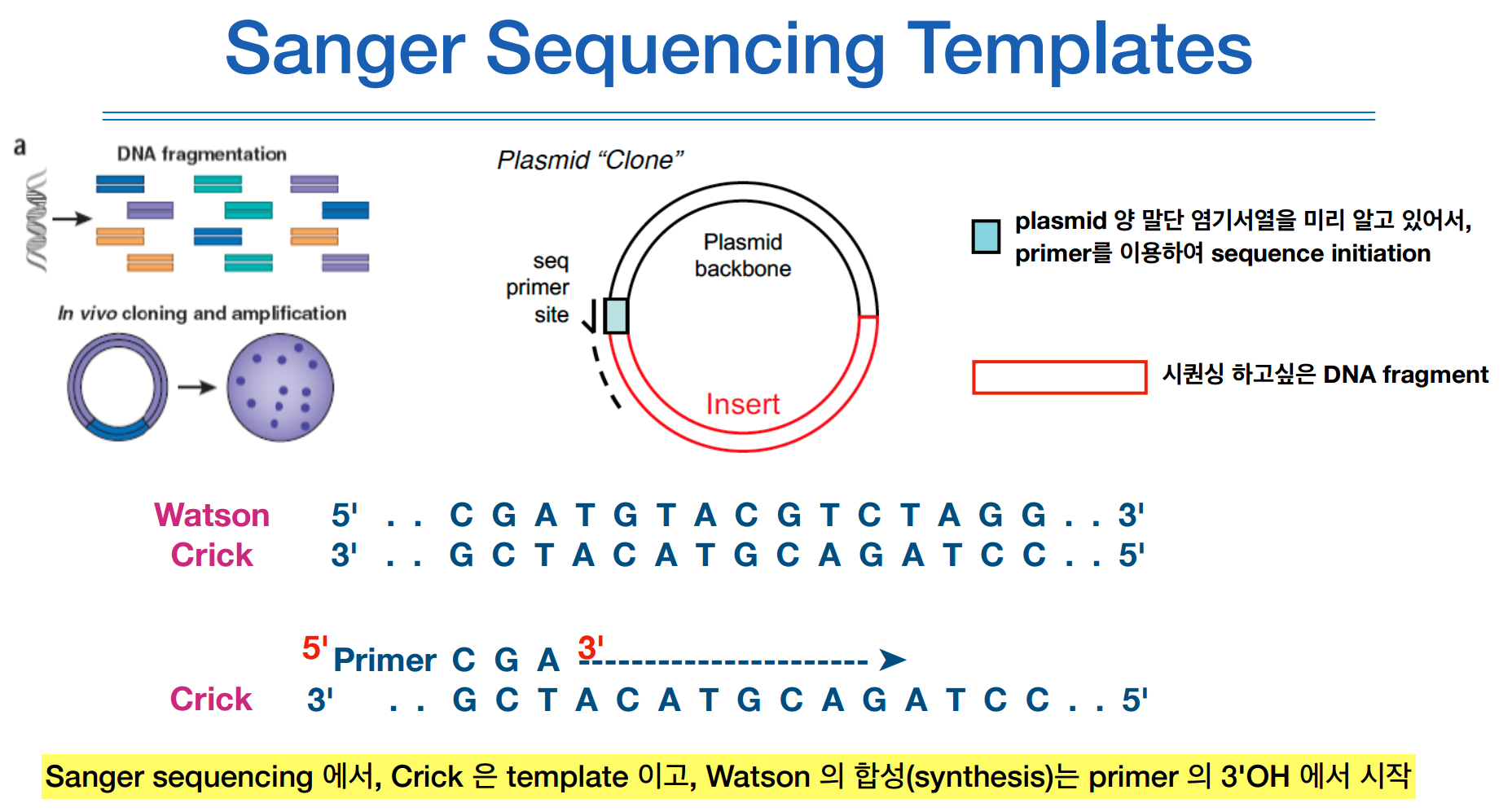
좌측 상단의 그림처럼 1970년에 개발된 restirction emzymes을 통해 DNA를 fragmentation할 수 있고,
박테리아는 저런 plasmid를 가지고 있는데, restirction emzymes를 사용하면 특정 위치를 자를 수 있다.
그런 다음 내가 가지고 있는 DNA fragmentation을 저기 안에 집어넣는다. 이를 transformation이라고 한다.
이렇게 집어넣고 박테리아를 다시 키우면, 박테리아가 자기 DNA와 앞서 넢은 fragmentation을 같이 증폭시킨다.
박테리아가 수천마리가 되면 나의 DNA fragmentation도 수천마리가 되는 셈이다.
증폭된 DNA를 뽑아내면 위 그림에서와 같이 watson crick과 같은 이중나선구조가 나온다.
이런 이중나선구조에 열을 가하면 분리가 되어 single strand가 된다.
여기에 primer를 붙히면 하나하나 합성이 되어간다.
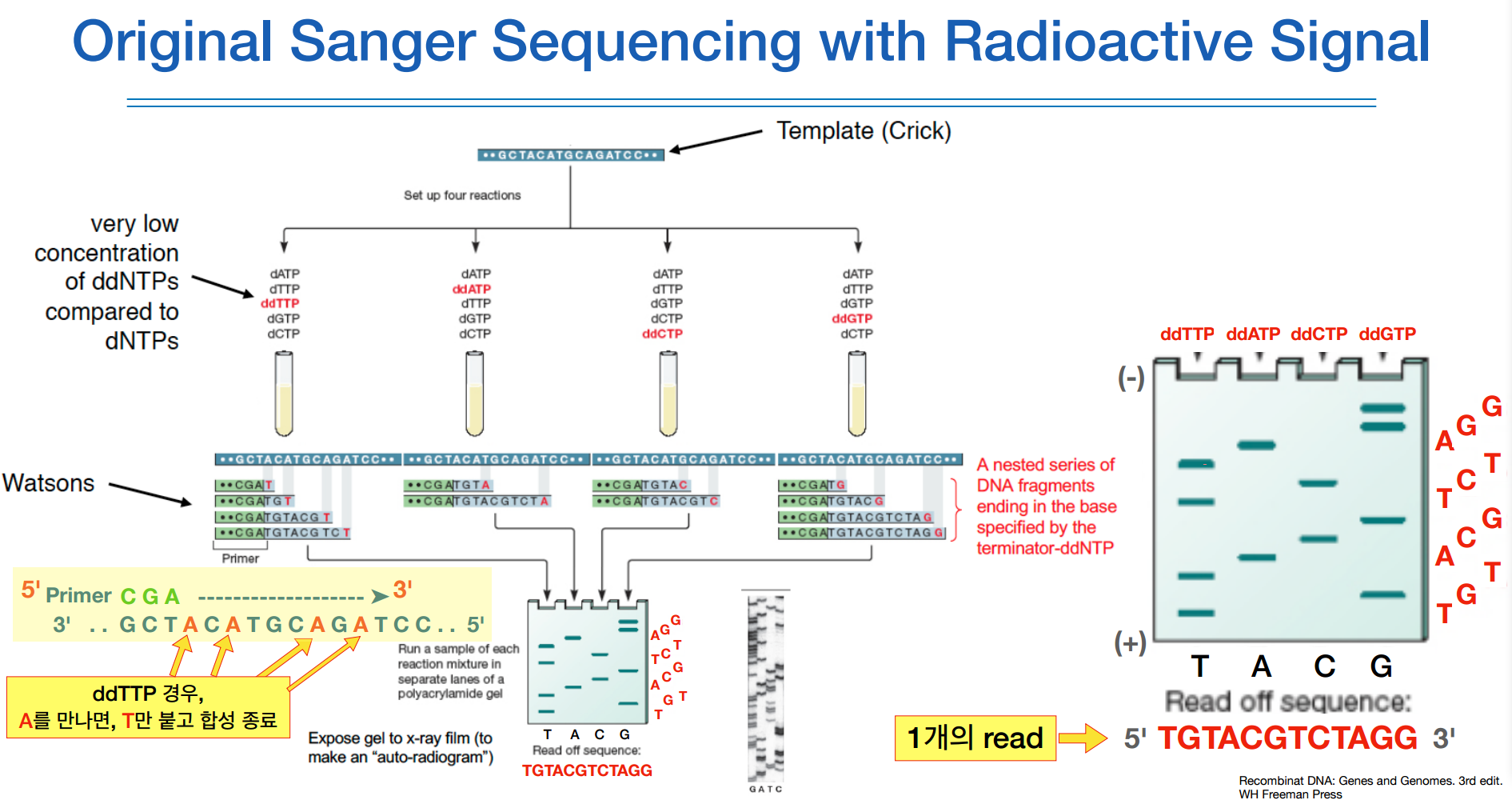
그림으로 살펴보면, 내가 가지고 있는 template를 각각의 ddNTP가 들어있는 튜브에 집어 넣는다.
ddNTP는 dNTP보다 저농도로 넣어준다.
그러면 각각의 튜브는 각각의 ddNTP가 붙어있는 염기서열에서만 termination이 일어난다.
그럼 다양한 길이를 가진 종결된 여러 조각들이 나오게 된다.
랜덤하게 합성이 일어나거나 종결이 되기 때문에.
이런 조각들을 다 꺼내어 각 튜브별로 전기영동을 하면, dna 조각들이 (-)에서 (+)로 가는 도중에 길이에 따라 짧은 것은 저항이 작기 때문에 멀리가고 긴 것은 멀리 가지 못하는 특징이 있다.
이를 이용해 우측 그림처럼 나오고, 짧은 애부터 읽어나가면 된다.
이것이 1개의 read 이다. 여기서는 1read에 12bp를 읽은 셈이다.
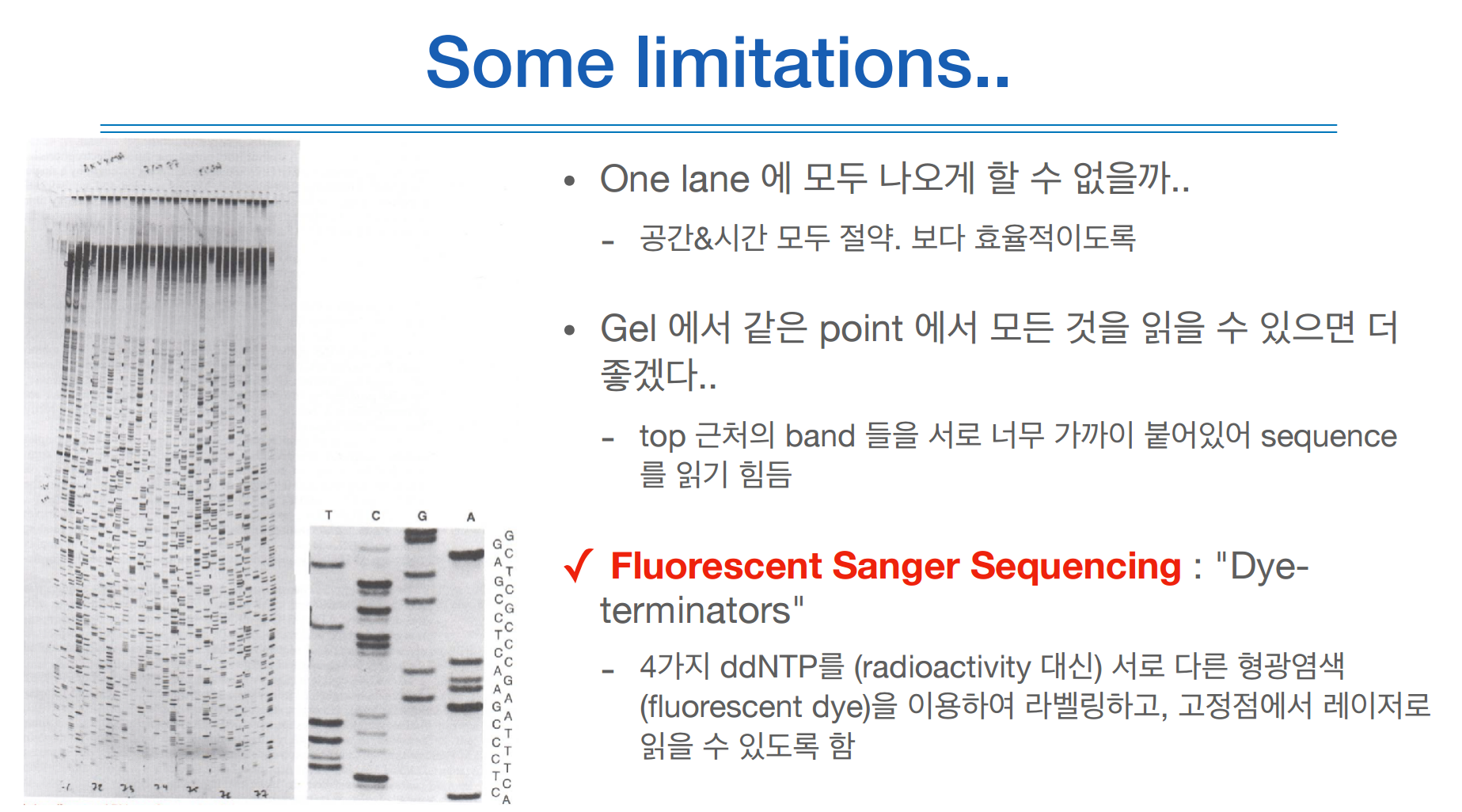
실제로 수행된 이미지를 보면 위와같다.
한계로는 너무 가까이 붙어있으면 읽기가 어렵다는 것. 너무 아날로그틱하다.
그래서 한개의 lane에 다 나오게 하고 싶다! 해서 아래와 같은 기술이 등장.
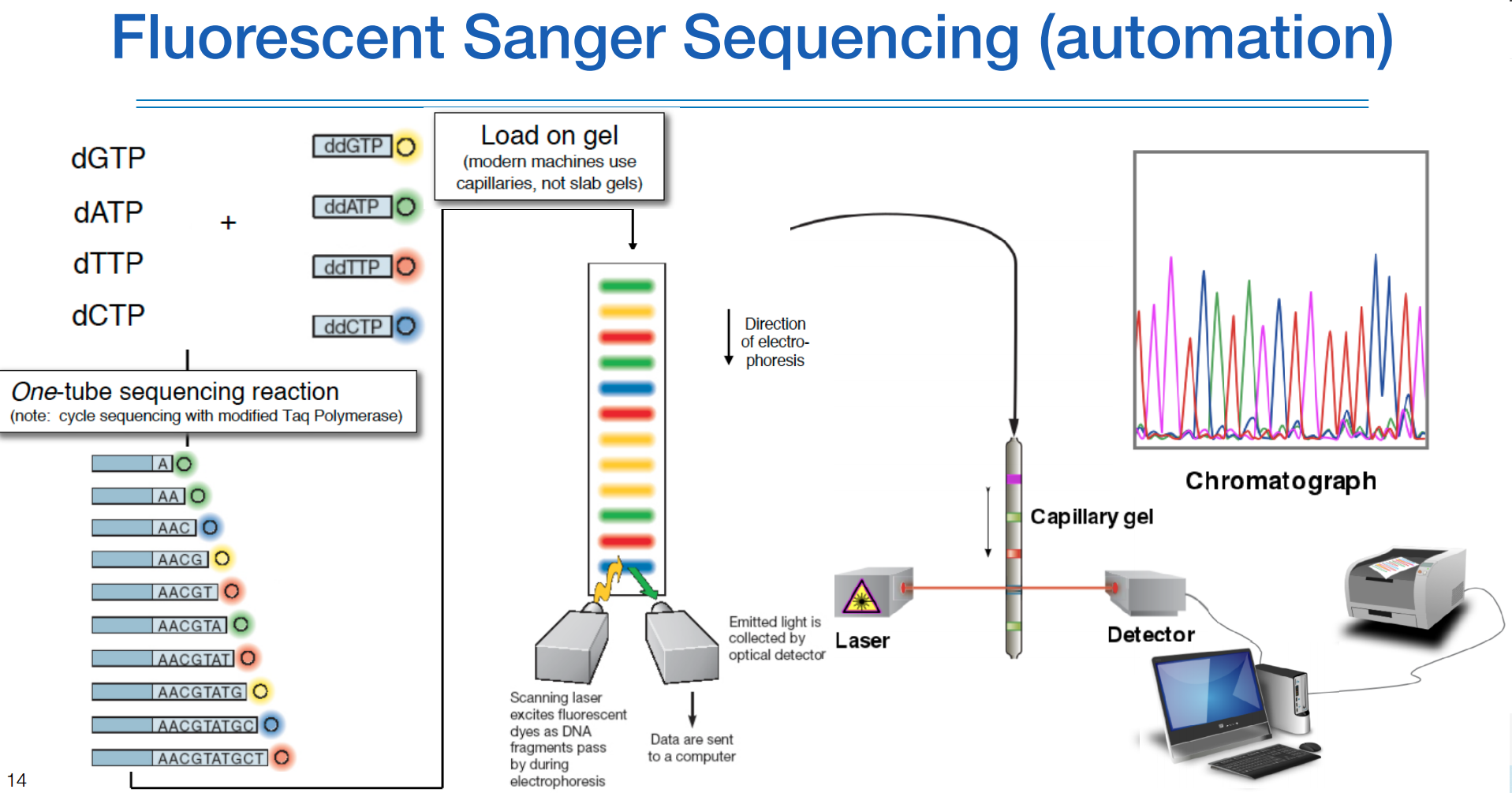
염기서열 마다 색깔을 다르게 하여 한 줄로 읽는게 가능하다.
현재 생어 시퀀싱이라고 하면 위와같은 것을 말하며, capillary 시퀀싱이라고도 한다.
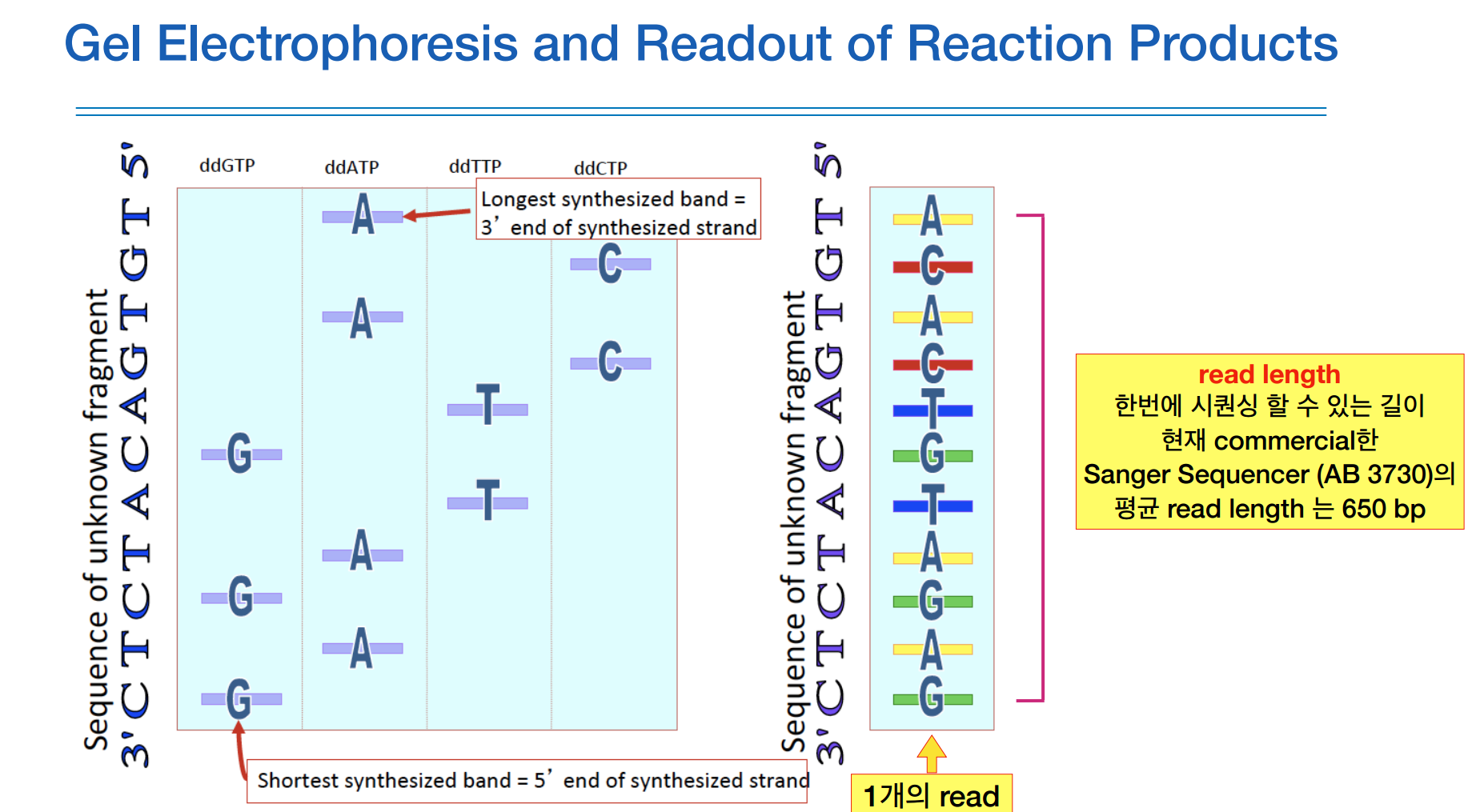
현재 생어시퀀싱으로 한번에 읽을 수 있는 read length는 650bp 정도 된다고 한다.

70~80년대는 1번에 12개의 리드를 할 수 있고, 1 read당 60bp정도 읽을 수 있었으니, 1런에 700bp정도 읽는다.
90~00년대는 기술이 더 발전하여 50kb/run이고, 00년대 이후로는 생어시퀀싱으로 100kb/run 정도 읽는다.

장단점은 분석이 정확하지만 읽는 길이가 짧다.
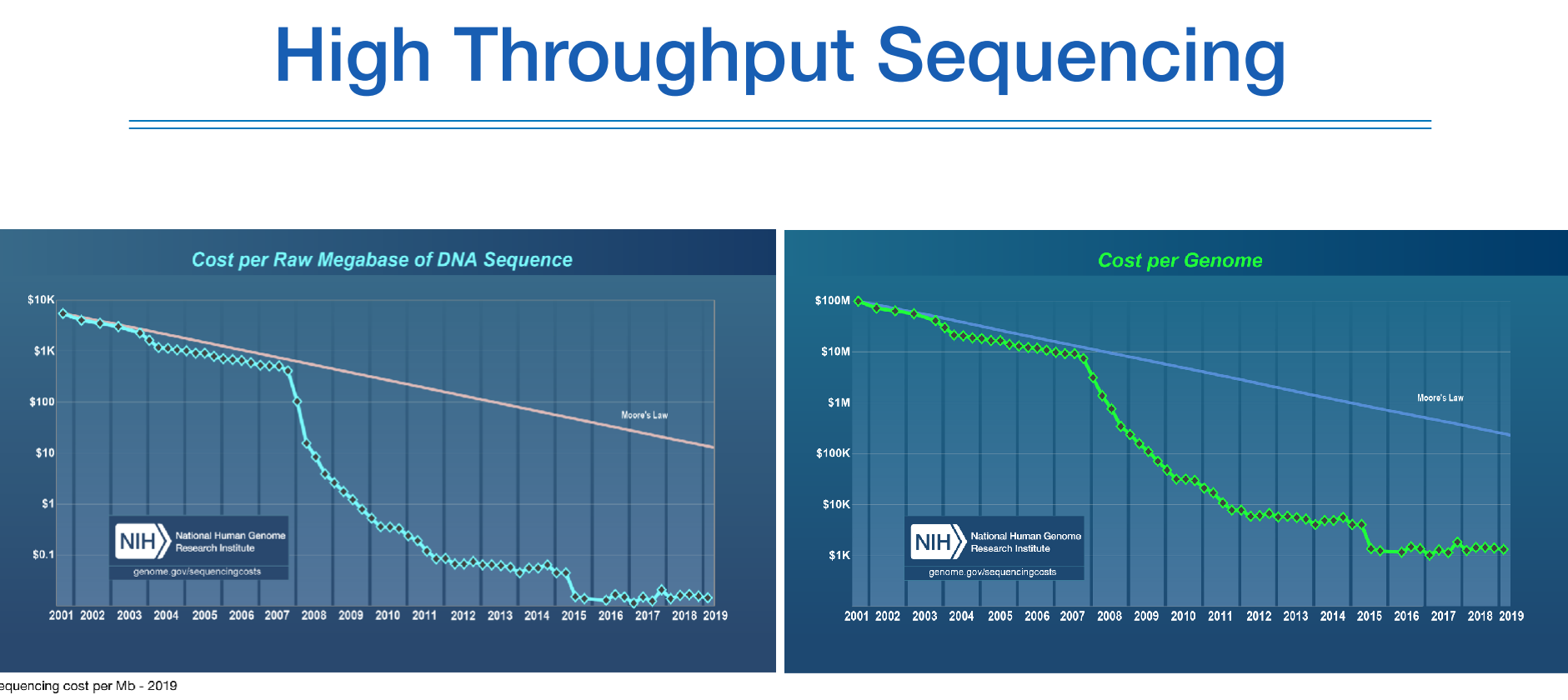
지놈 프로젝트는 바로 생어시퀀싱으로 이루어졌다.
한편, 2006년 NGS의 등장으로부터 노하우가 축적이 되면서 07년서부터 급격하게 시퀀싱 비용이 저렴해진 것을 알 수 있다.

NGS는 생어에 비해 정확도는 떨어지지만 high throughput 시퀀싱으로 많은양의 처리를 할 수 있다.
그에 따라 가격도 저렴하다.
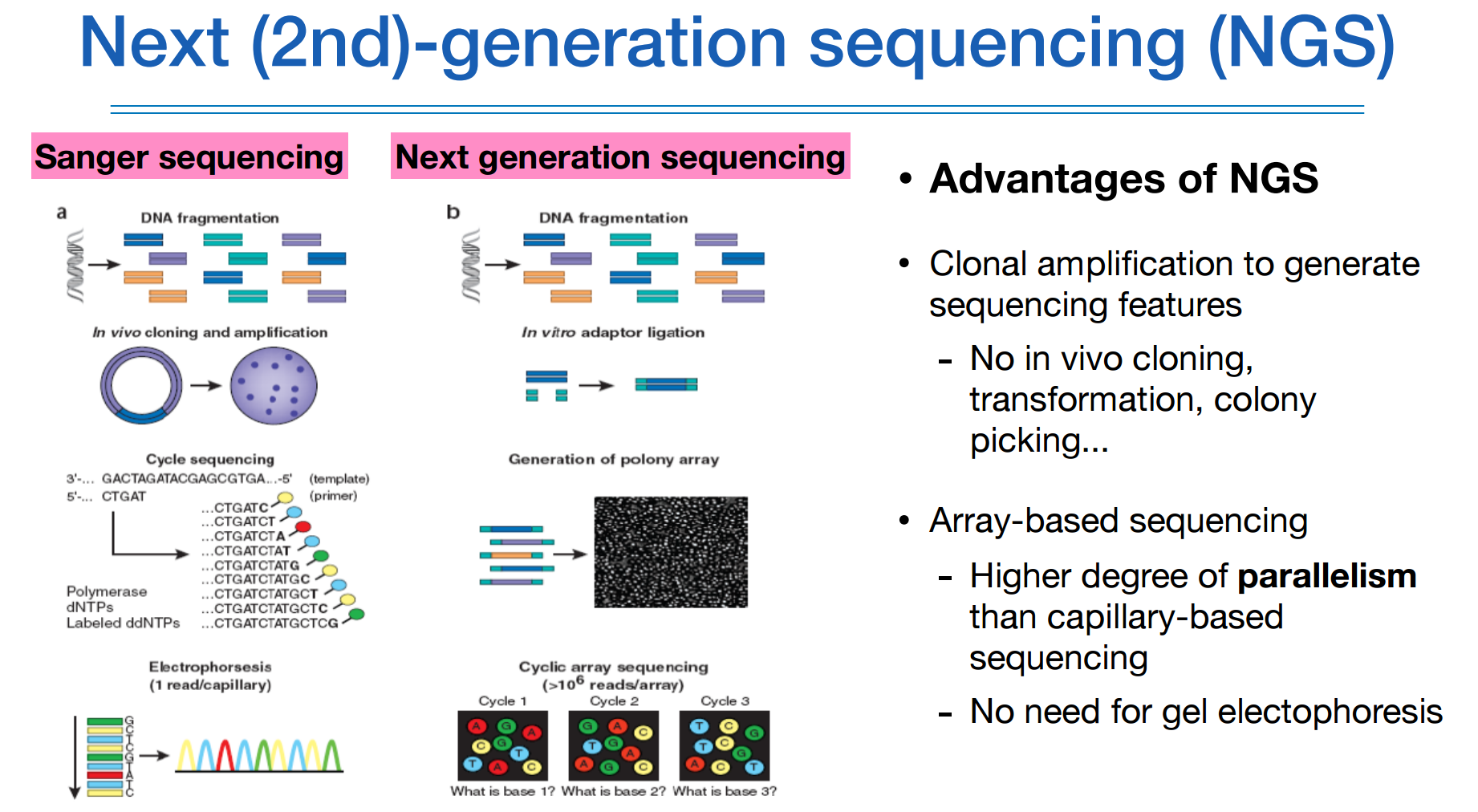
생어시퀀싱이나 NGS나 모두 fragmentation을 하지만 기술이 발전하면서 길이가 길어졌다.
amplication도 PCR을 통해 비교적 간단히 할 수 있으며, 한조각 한조각하는것이 아니라 모든 fragmentation을 한번에 massive하고 parellel하게 amplication할 수 있다.
또 바로 동시에 시퀀싱을 할 수 있다.
전기영동을 하지않고 이미지처리 기술을 이용하여 한번에 처리한다.
이 두 방법은 원리는 유사하나 얼마나 많은 양을 간편하게 처리할 수 있느냐의 차이이다.

흐름은 다음과 같다.
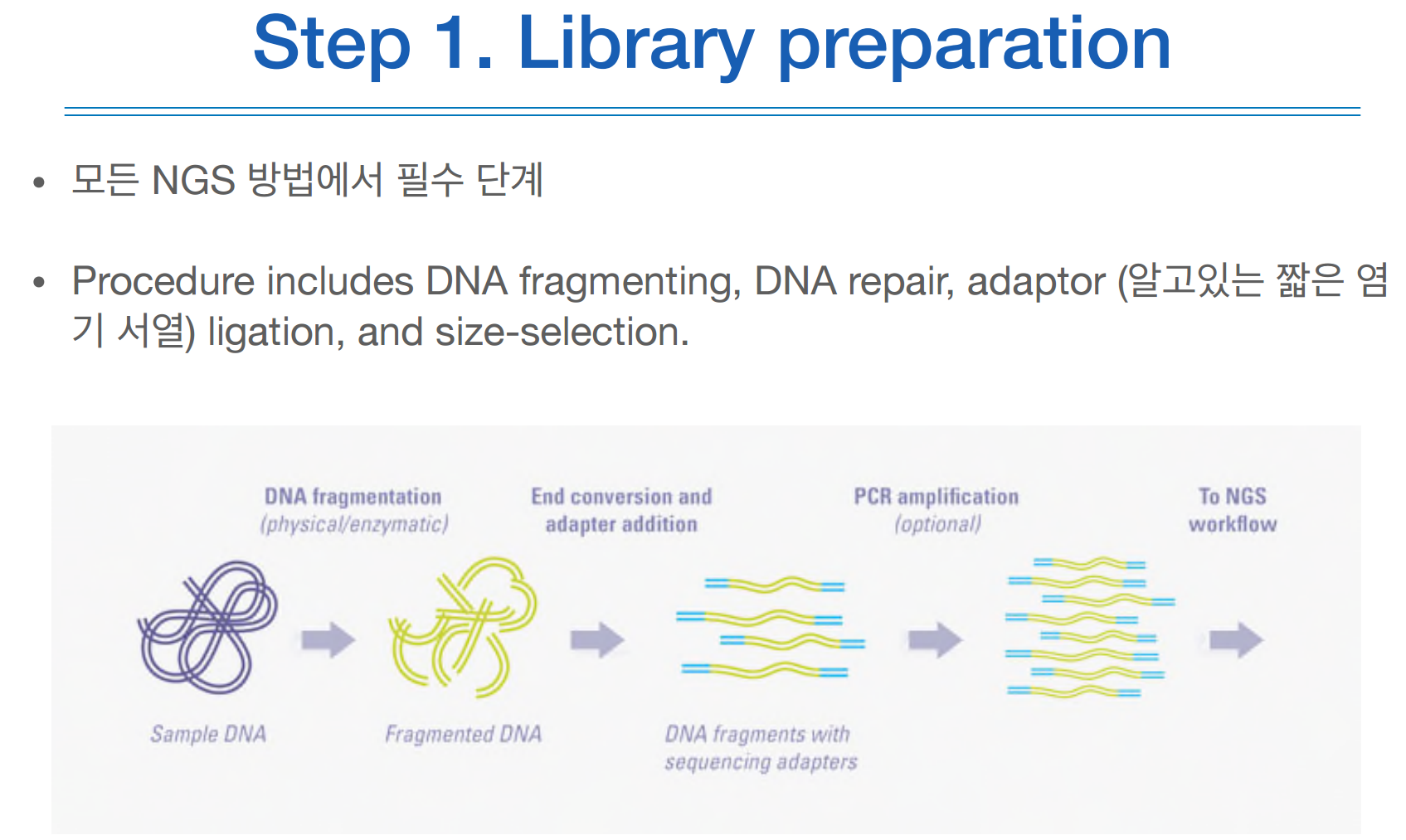
1) library라는 adaptor를 DNA 조각 양 끝에 붙혀서, 어떤 재료판과 짝꿍이 되어 붙을 수 있게 한다.
2) PCR을 이용해서 amplication
3) 시퀀싱과 이미징

요즘에는 emulsion PCR보다 bridge PCR을 많이 사용하여 일루미나 머신을 가장 많이 사용한다.
그래도 아직까지 사용하기는 해서 emulsion 방법을 간단하게 보면,
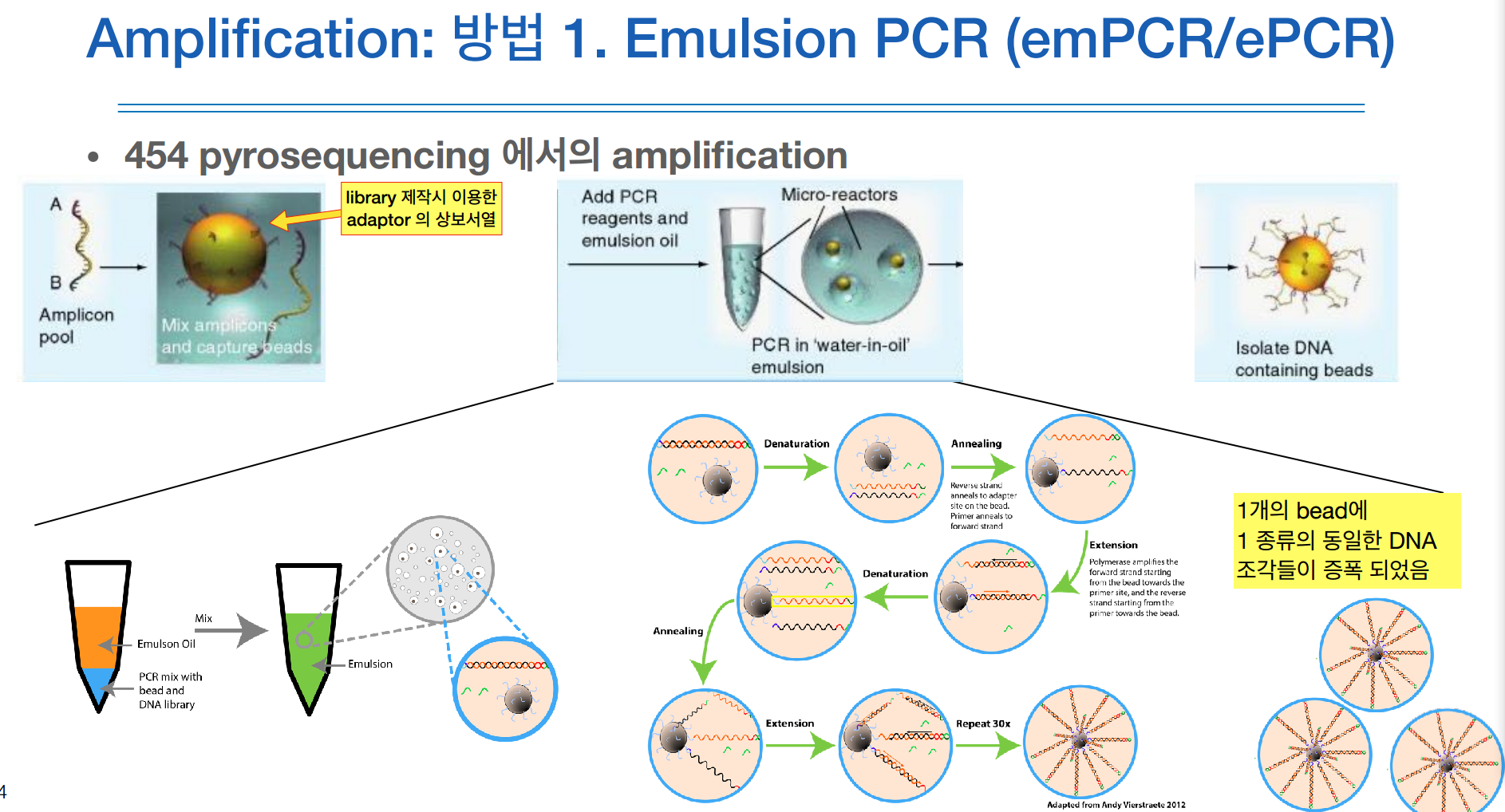
작은 beads(비즈)에 adaptor의 상보서열을 붙혀놓았다.
한 튜브에 DNA polymerase를 넣고 거기에 추가로 emulsion oil을 같이 넣는다.
튜브에 어떤 강한 자극을 주게 되면, 순각적으로 물과 기름이 섞이면서 기름이 비즈를 감싼다.
이것이 이멀젼 상태.
그럼 이 상태는 오일 하나하나마다 하나의 비즈가 들어가서 PCR이 진행된다.
PCR이 끝나면 모든 비즈가 같은 DNA로 복제가 된다.
그럼 우측 하단 그림처럼 증폭이 됨.
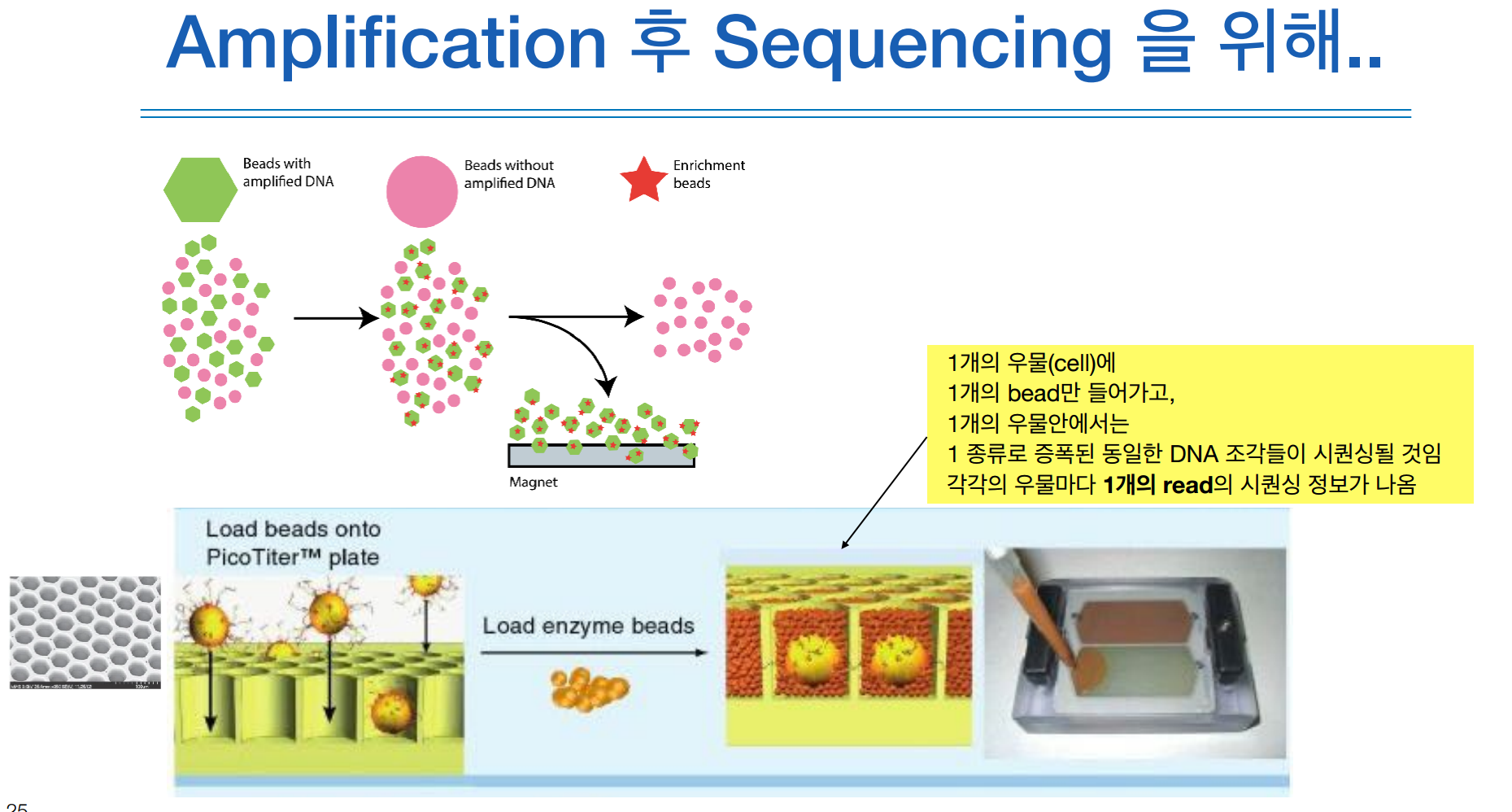
그럼 magnet 판에 비즈 하나씩만 들어같 수 있는 작은 홈이 있는데 거기에 비즈를 집어넣고, 비즈가 나오지말라고 더 작은 비드로 매꿔준다.
이 상태가 amplication이 끝나고 시퀀싱을 준비하고 있는 상태이다.
이 한판을 시퀀서에 들어가면 한판으로 시퀀싱 데이터가 나온다.
또 다른 amplication 방법은 (일루미나에서 사용하는 방법)
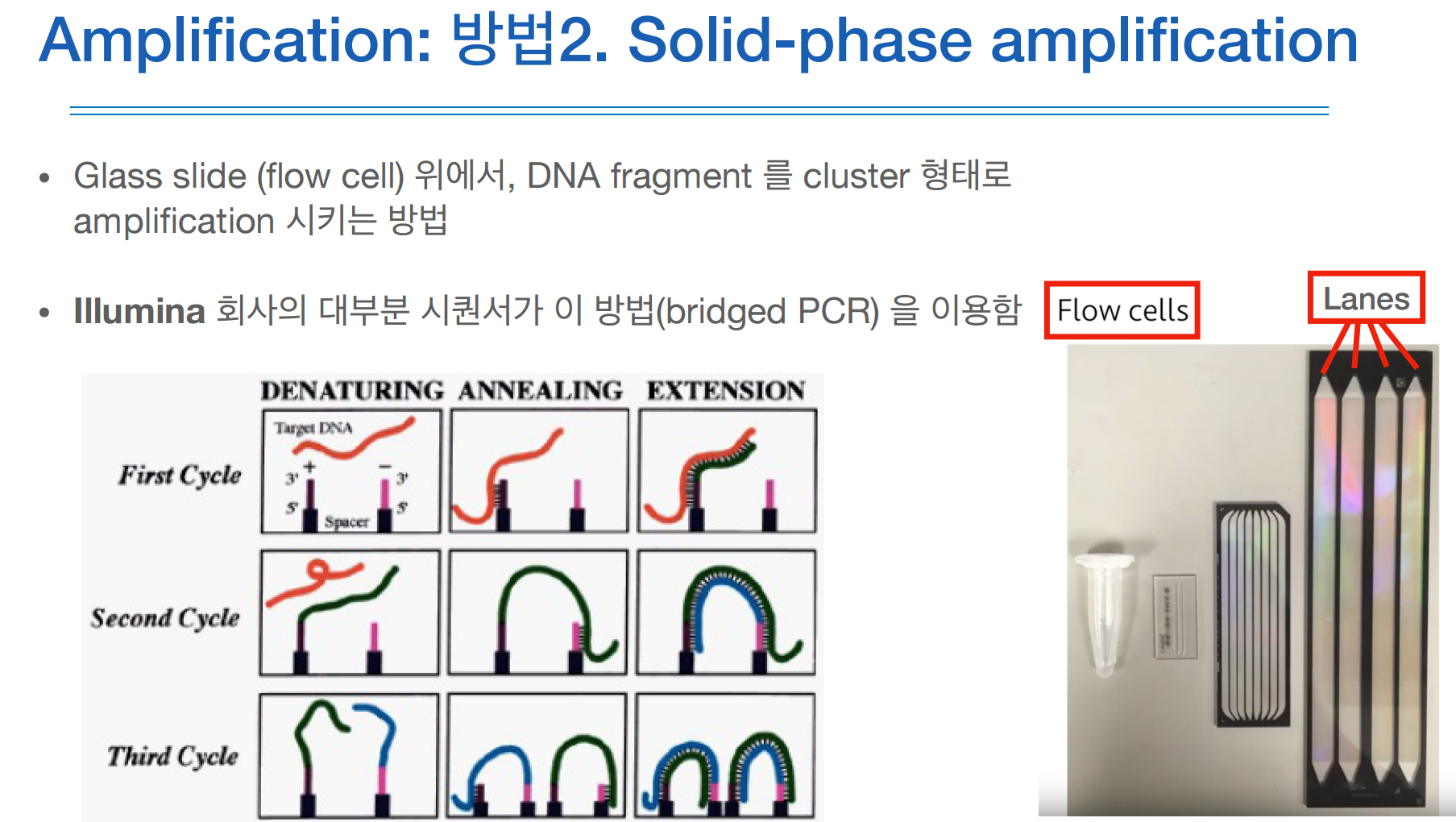
이멀션처럼 어떤 용액상태에서 되는게 아니고, 어떤 고체의 판 위에서 되는거라 솔리드 페이즈라고 부르고,
이 용어보다는 브릿지 PCR을 더 많이 사용한다.
그 이유는 증폭이 되는 과정에서 위 그림처럼 어떤 다리를 형성하는 듯한 모양세를 띄기 때문이다.
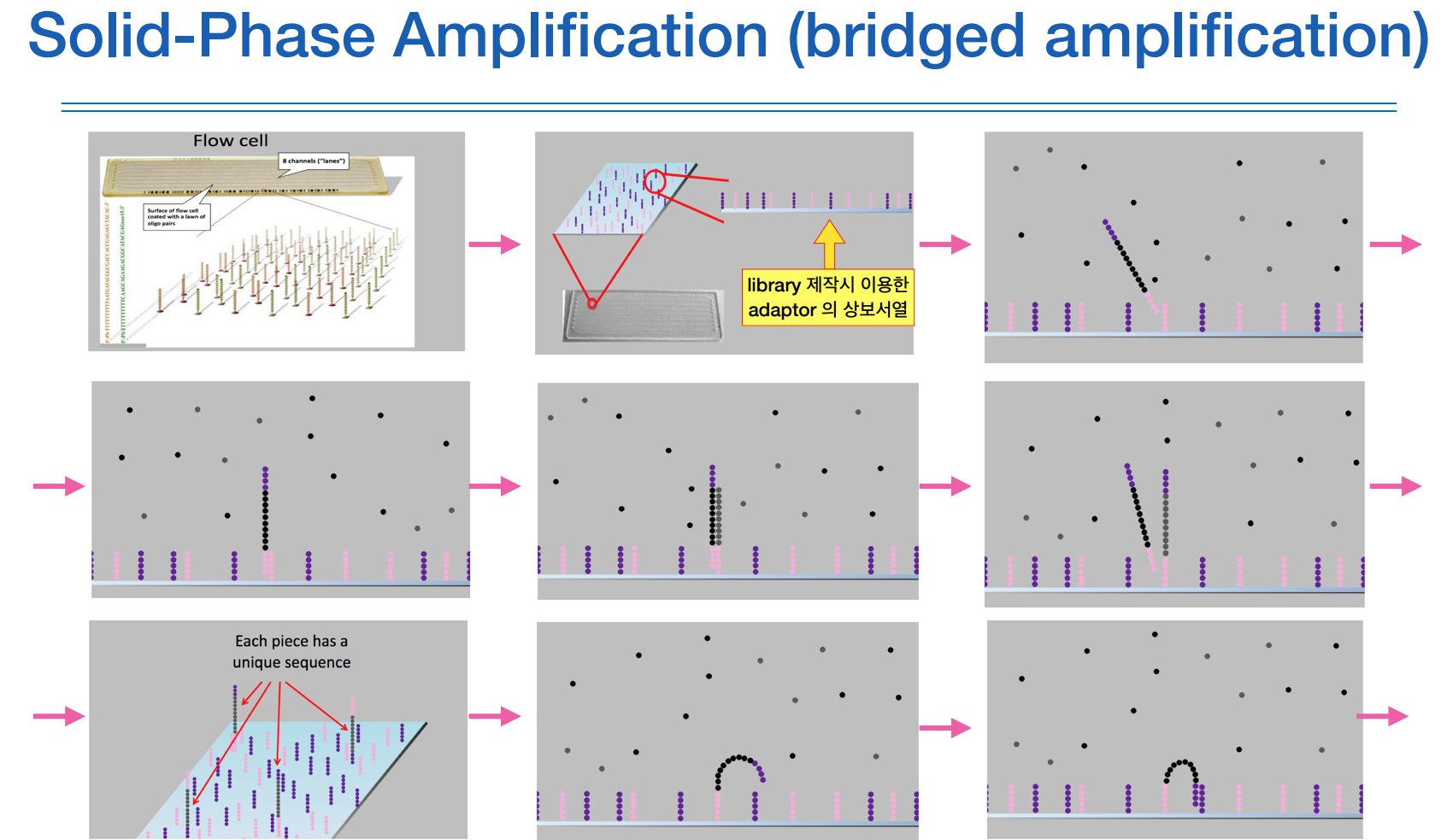
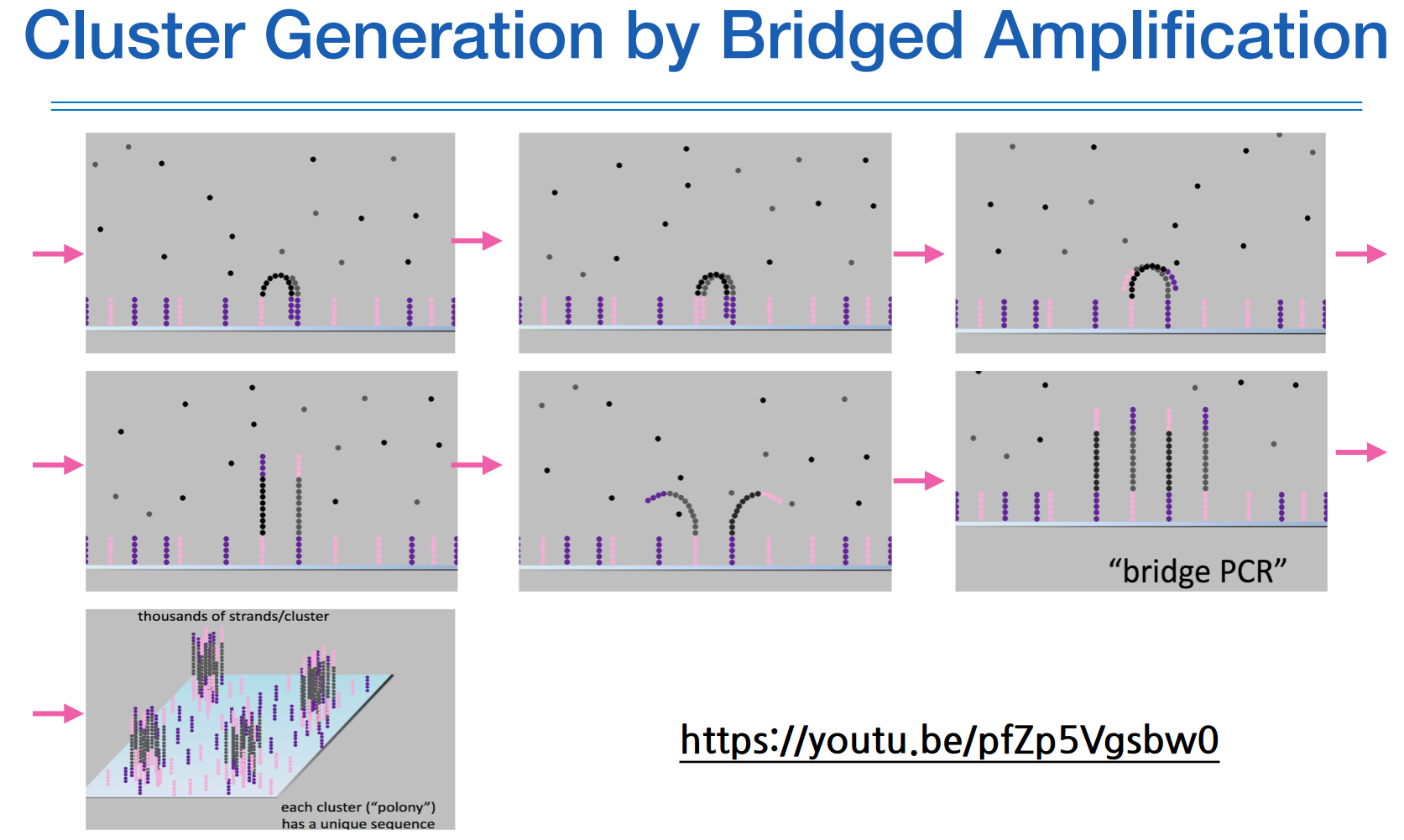
유리판위에 adaptor의 상보서열이 붙어있고, 나의 DNA 조각들이 거기가서 붙으며 증폭이 된다.
그럼 최종적으로 증폭된 DNA조각마다의 어떤 클러스터를 형성한다
한 클러스터가 하나의 read 단위가 된다.

시퀀싱하는 방법은 SBS, SBL 뭐 등등이 있다.
454머신은 현재 사용하고 있는 머신은 아니지만, 그 머신이 사용했던 SBS방식 중 pyrosequencing 방식은 현재도 사용하고 있다.
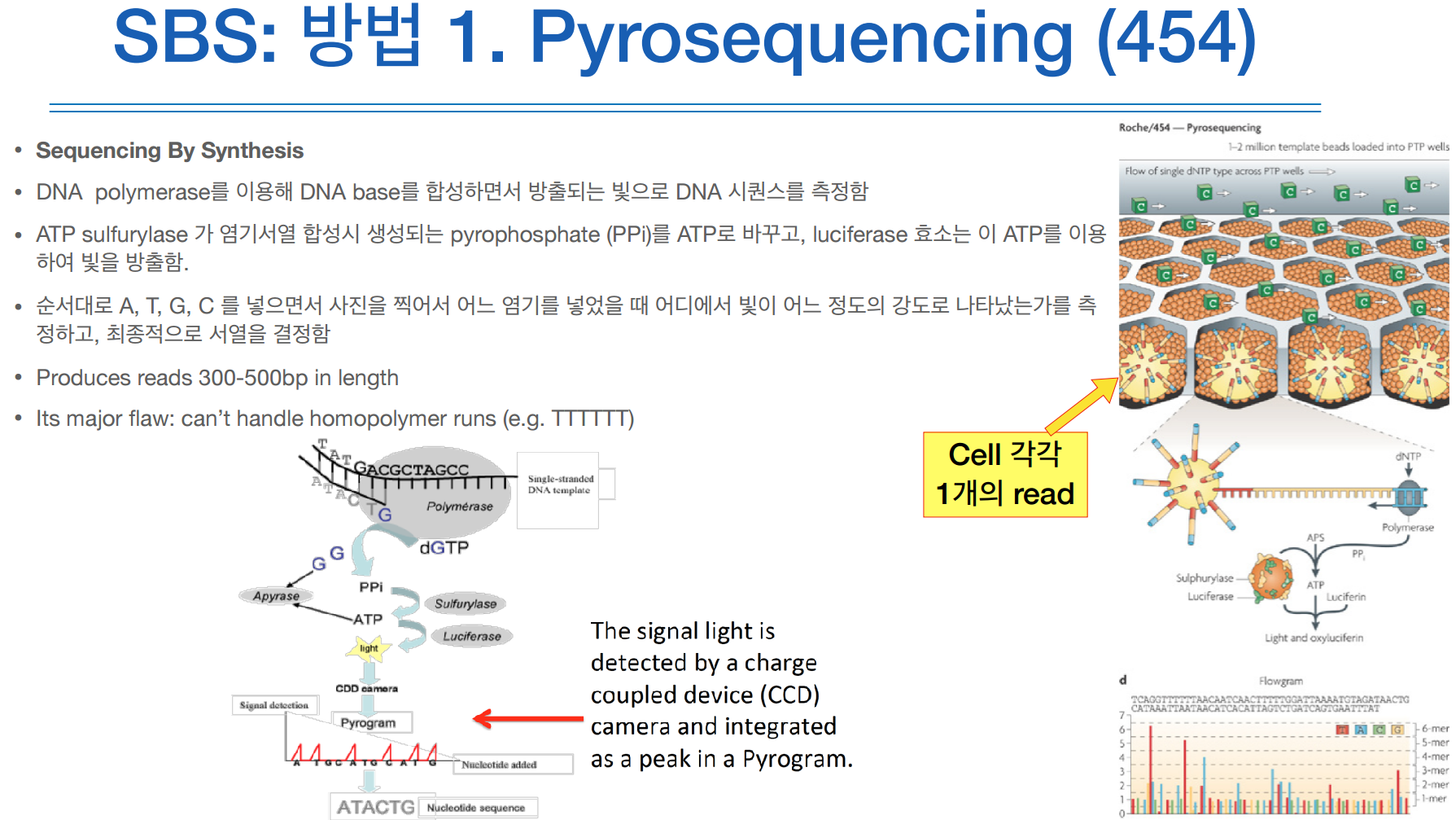
파이로시퀀싱은 이멀젼 PCR을 하고 사용을 한다.
DNA를 합성하면서 시퀀싱을 하기 때문에 SBS라고 불린다.
합성을 하려면 항상 DNA polymerase랑 dNTP가 필요하다.
이때 여기서는 ATP sulfurylase랑 luciferase라는 걸 추가로 집어넣는다.
그러면 ATP sulfurylase 가 염기서열 합성시 생성되는 pyrophosphate (PPi)를 ATP로 바꾸고, luciferase 효소는 이 ATP를 이용하여 빛을 방출한다.
키포인트는 DNA를 합성하면서 빛을 발생시켜 그 신호를 처리하여 읽는 다는 것이 핵심이다.

이 방법은 역시 SBS방식인데 생어 시퀀싱처럼 터미네이션을 이용한다.
하지만 완전히 종결되는것이 아니라 거기서 또 다시 시작하는 리버서블 터미네이션 방식이다.
합성하고 -> 형광물질이 반응하고 -> 다시 합성하고 이 순서를 반복한다.
ddNTP를 넣지않고 리버서블 터미네이터가 붙어있는 dNTP를 넣는다.
그림으로는 아래와 같다.
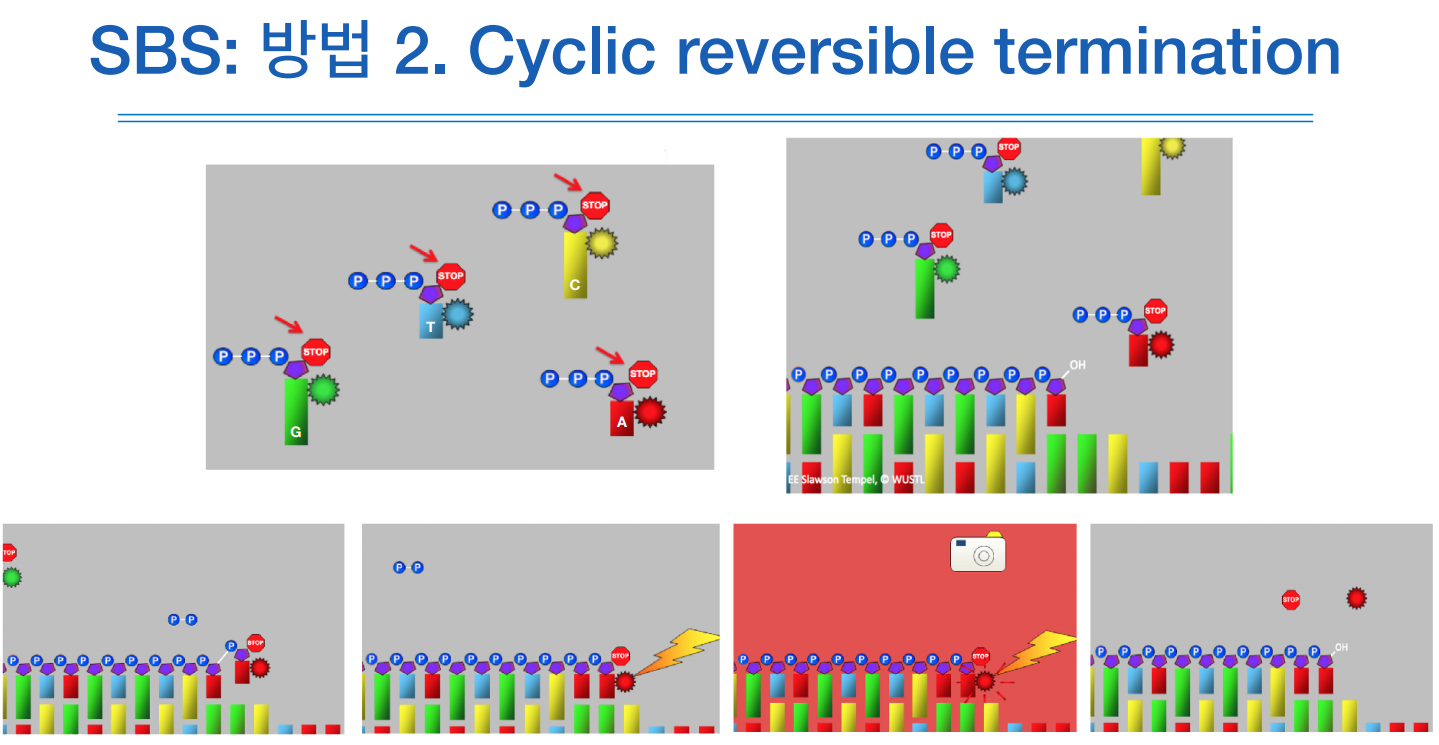
위 그림처럼 OH에 dNTP가 붙지마나 dNTP에 stop이라는 블로커가 붙어있어서, 합성되면 반짝하고 이를 광학 카메라로 사진을 찍고, 워싱을 하면 stop 브로커가 떨어지고, 그럼 일반적인 dNTP가 되어 다시 합성이 진행된다.
다른 그림으로 보면 아래와 같다.
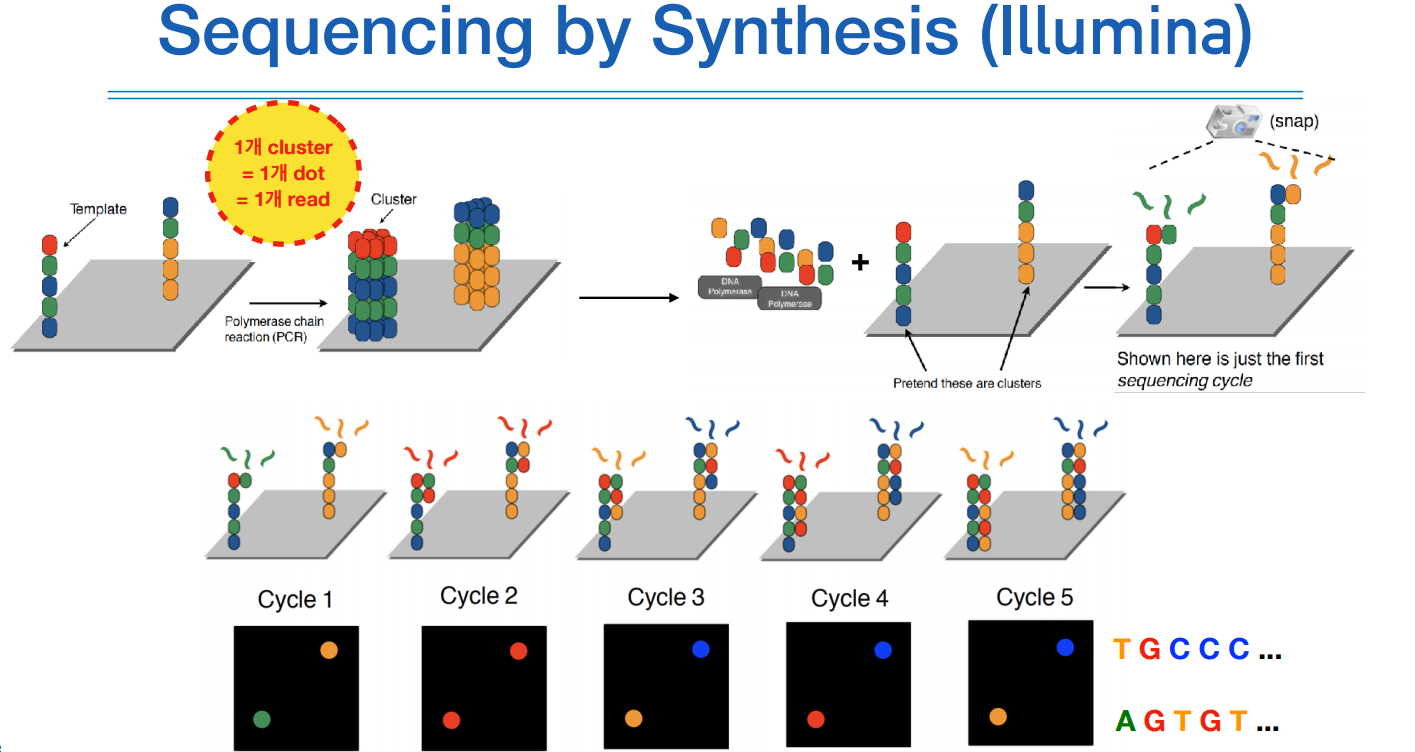
read가 20bp면 사이클이 20번 돌아서 사진이 20장이 나온다.

위 그림처럼 이미징하는 장비가 굉장히 고가이고 크기 때문에, 이미징 기술이 있는 시퀀싱 장비면 장비도 크고 비싸다.
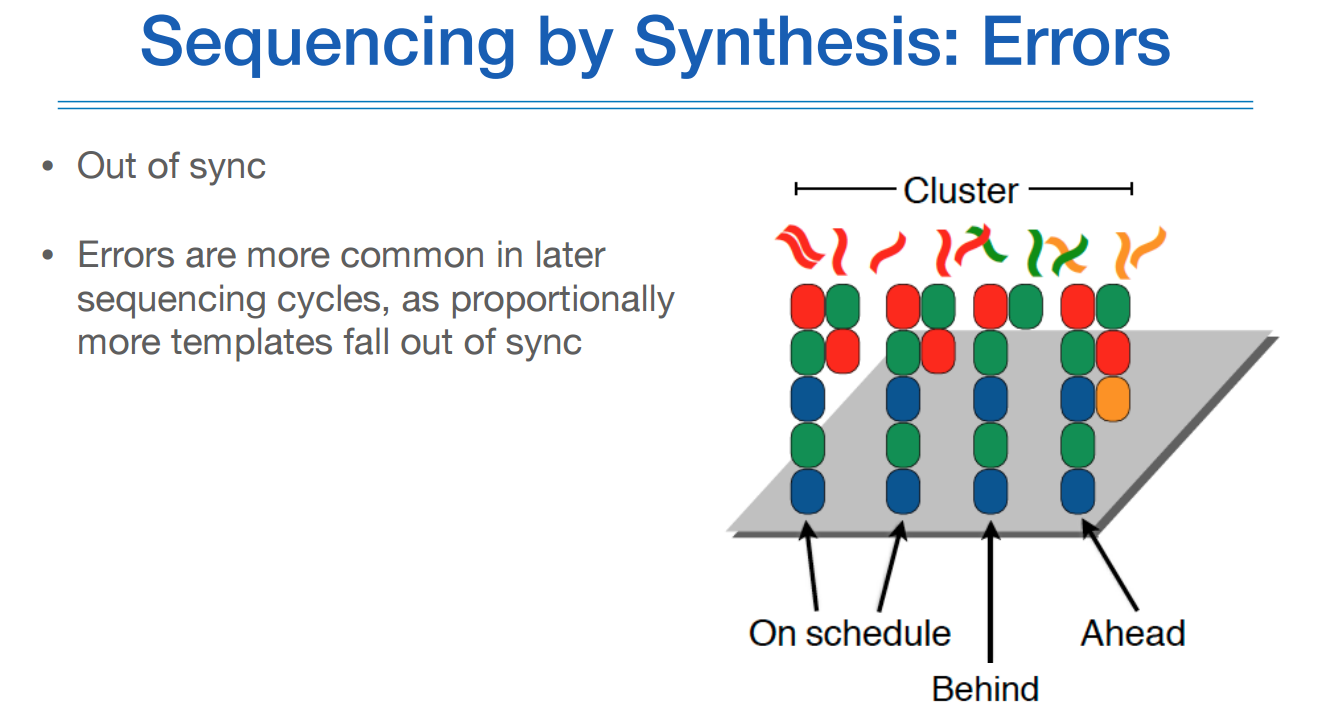
SBS방식의 에러는 위와 같다.
합성이 동시에 순차적으로 잘 되면 좋겠지만, 어느것은 보다 느리게 합성되거나, 빠르게 합성되는 것이 있다면 하나의 클러스터에서 발생하는 불빛이 일정하지 않을 수 있다는 점이다.
이것이 바로 아웃오브싱크이다.
그래서 read length가 너무 길면 처음에는 퀄리티가 좋다가 나중에는 퀄리티가 안 좋아진다.
그래서 롱리드시퀀싱이 어려운 점이 있다.

일루미나 홈페이지에 가면 엄청나게 많은 시퀀싱 장비들이 있다.
나누는 기준은 쓰르풋차이이다.
30Gb이면 10명정도를 동시에 할 수 있다. (1명당 3Gb(기가베이스페어)이기 때문에)
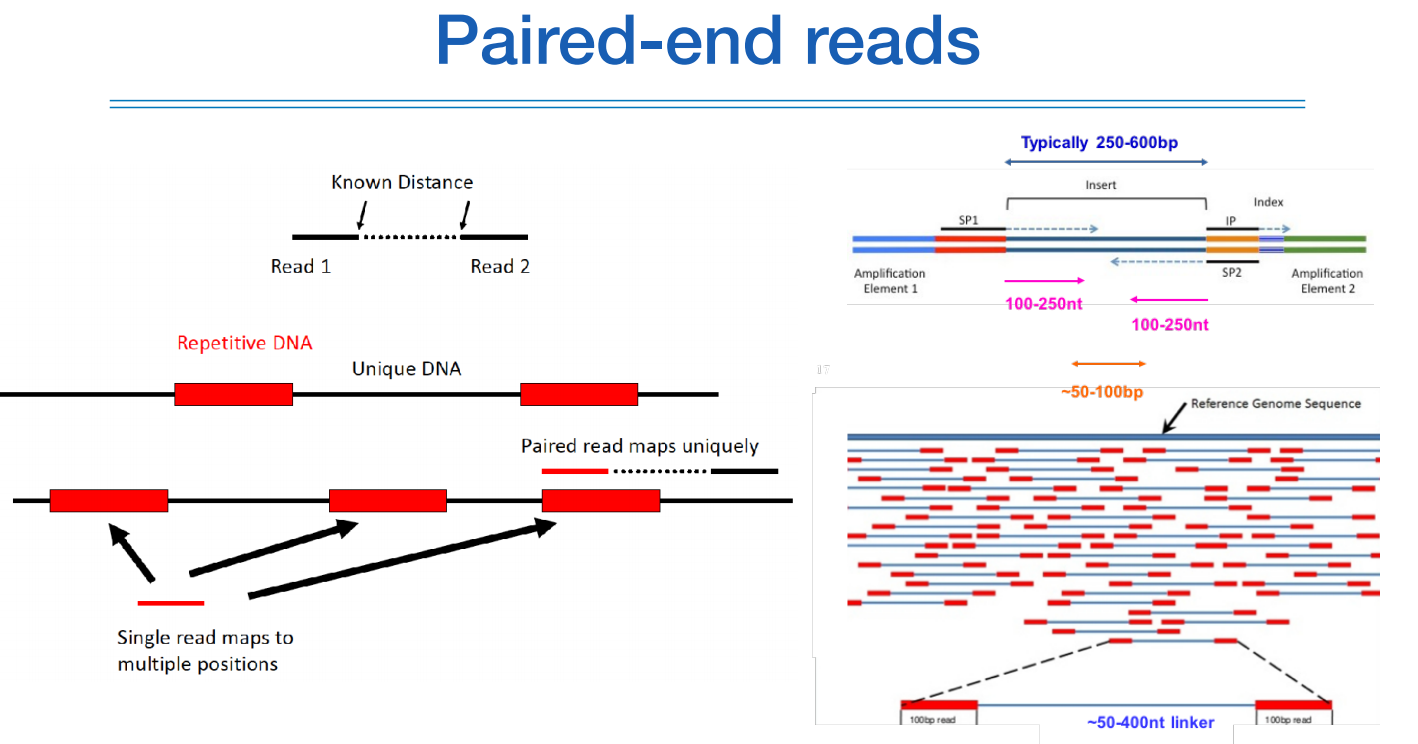
paired end read를 하면 하나로 통합하는 매핑과정을 통해 정확도를 올릴 수 있다.
시퀀싱을 하면 하나의 single read로 존재하는데 얘는 보통 human genome reference를 통해 어느 위치에 있는지 매핑을 한다.
근데 짝궁 read가 있으면, 얘네 둘은 본래 서로 얼만큼 떨어져있는지 알고 있기 때문에 하나의 read의 위치만 알면 다른 하나의 위치는 자동으로 정해지는 그런 원리이다.
현재 이러한 일루미나 방식이 가장 많이 사용하는 방식이다.
그 이유는 정확도, 가성비 등 종합적인 이유에서이다.

그럼 SBL 방식은 무엇이냐? ligation을 이용하는 것이다.
라이게이션은 효소의 작용을 통해 두 개의 핵산 단편을 연결하는 것이고, 이 DNA ligase를 사용해서 3` -> 5`으로 DNA 조각을 붙혀나간다.
하지만 고비용이라 현재 사장된 방식이다.

시퀀싱 방식 중 이 세번째 방식은 이미징을 사용하는 방식이 아니라 2.5 세대라고도 한다.
이미징하지 않기 때문에 장비 크기도 작고 비용도 저렴한 편이다.
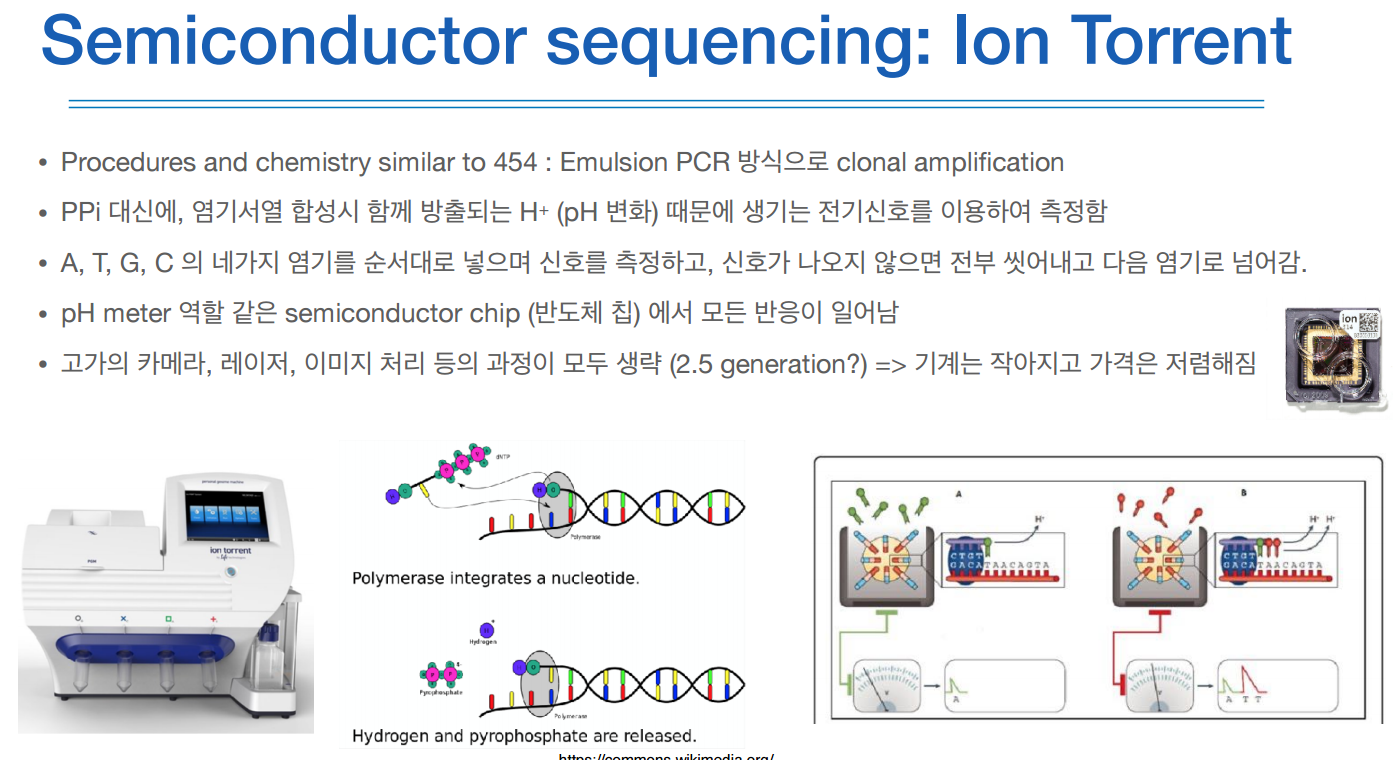
이 방식은 amplication은 emulsion PCR 방식으로 한다.
하지만 여기서는 PPi 대신에, 합성될때 함께 방출되는 프로톤(H+)에 의해 생기는 전기신호를 측정한다.
염기서열마다 나오는 프로톤이 조금 달라서 pH가 변한다고 한다.

이 3세대의 특징은 amplication이 없다는 것이다.
amplication의 목적은 강한 신호를 얻기 위함인데, 이를 어떻게 해결했을까?
증폭이 아니라 단일 molecules로 시퀀싱한다고 해서 SRMT[스마트, Single molecule real time sequencing] 시퀀싱이라고도 한다.
종류는 크게 두가지로, SBS가 필요한 PacBio와 SBS도 필요없는 Nanopore가 있다.

PacBio는 어떻게 시퀀싱을 할까?
3세대긴 한데 여전히 이미징을 하기때문에 잘 안쓰긴 한다고 한다.
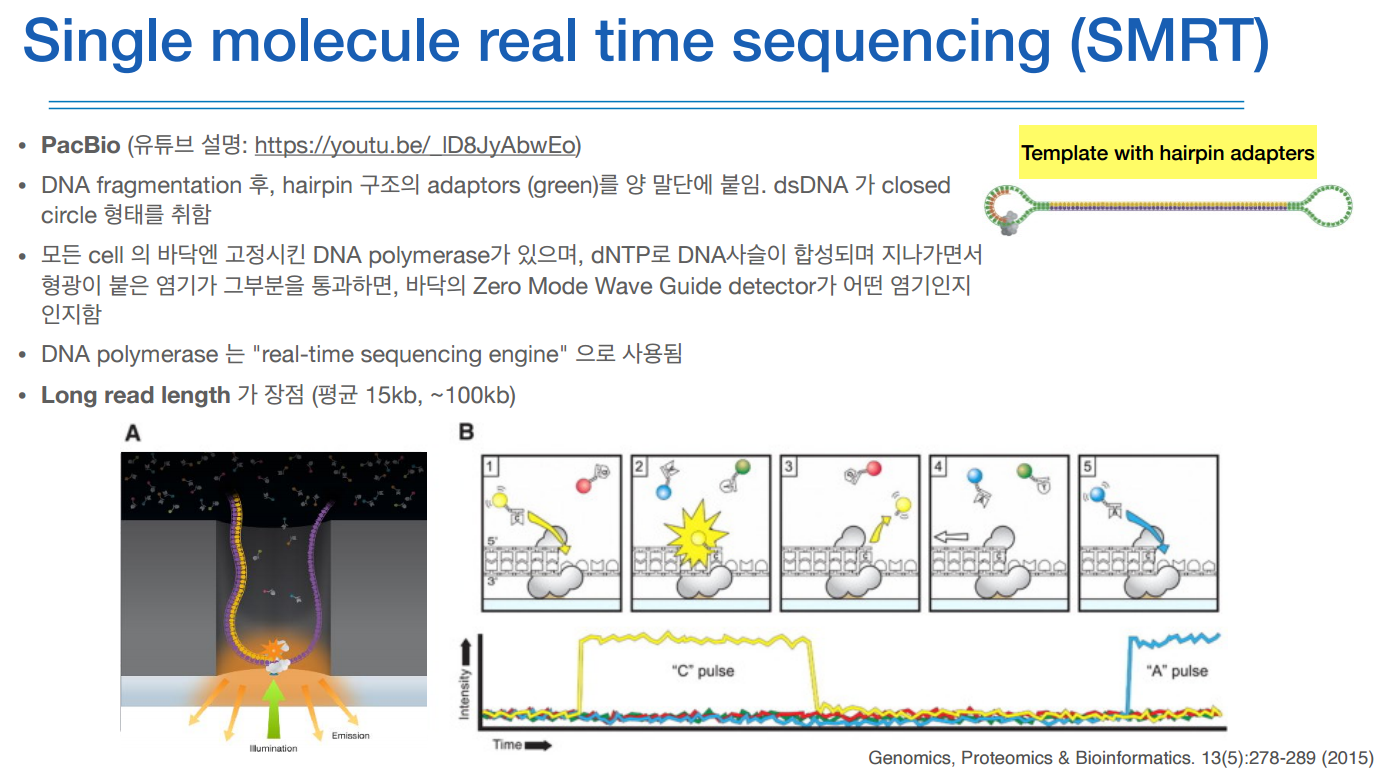
PacBio의 DNA fragmentation은 특이하게 헤어핀 구조이다.
DNA fragmentation에 어댑터를 붙힐때 열을 가하여 벌어지게 한다음 어댑터를 붙혀 링 형태로 만든다.
이 형태를 팹바이오 시퀀서 셀에 넣으면 A와 같은 형태로 들어가고 (싱글 strand 형태), 그 셀 안에는 광학이미징을 엄청나게 세게 하여 디텍팅하는 고가의 장비가 있다. (Zero Mode Wave Guide detector)
또 다른 특징으로는 헤어핀 구조이기 때문에 끊임없이 반복해서 측정할 수 있다. 그리하여 정확도를 높힐 수 있다.
따라서 롱 리드가 가능하다는게 장점!

마지막으로 나노포어는 이미징이 없다.
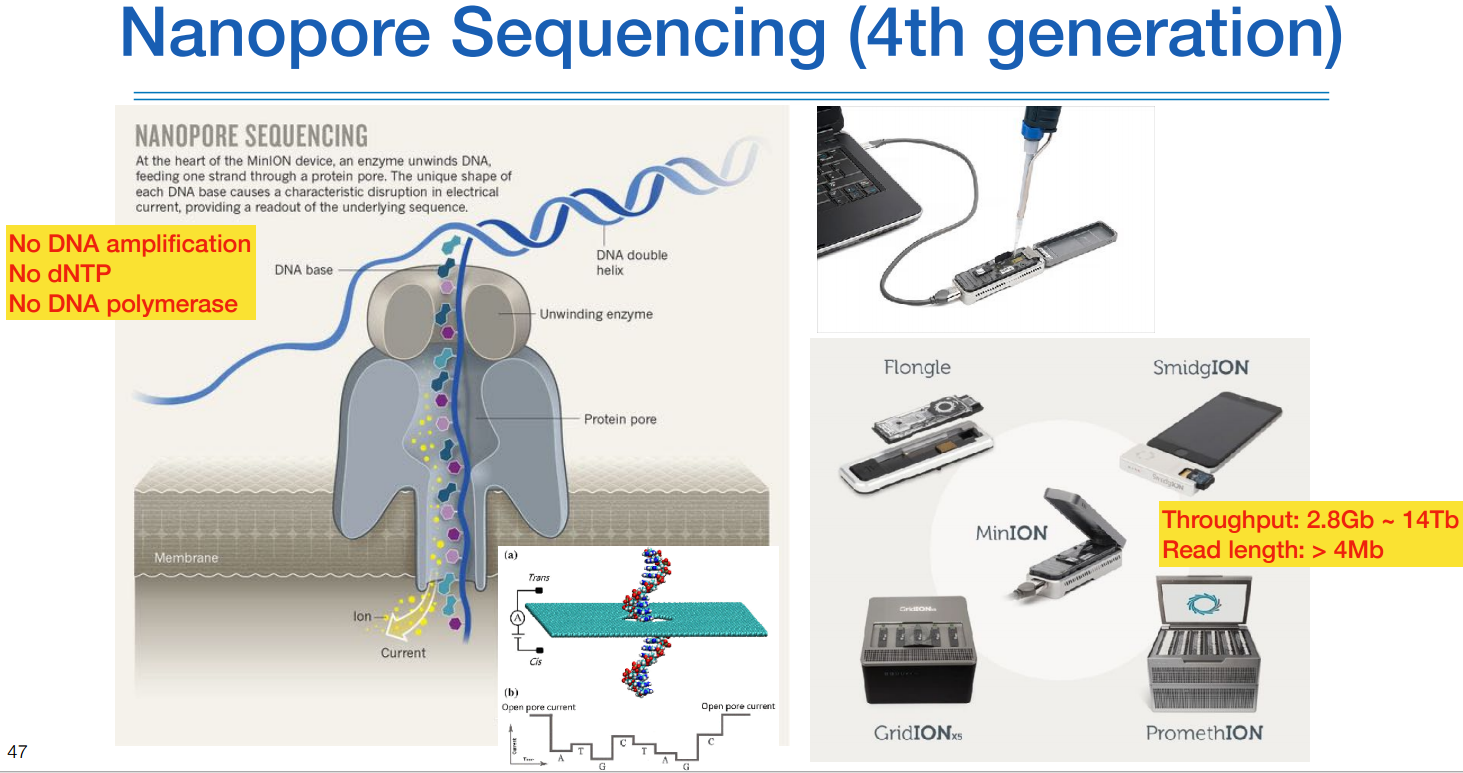
그리하여 위와같이 유에스비같이 엄청나게 작다.
하나의 모듈의 쓰르풋은 작지만 여러개 붙혀서 크게 만들수도 있다. (우측 하단 그림)
어떻게 하냐면,
멤브레인(Membrane, 얇은 막)에 전류를 계속 흐르는 상태에, 싱글 strand DNA 가닥을 그대로 계속 흘려보내준다.
(통째로는 안되고 역시 fragmentation)
어댑터를 붙힌 DNA를 인식할 수 있는 어떤 장비로 인해 DNA가 전류차에 의해 쪼로록 들어가게 된다.
염기서열마다 다른 전이차를 이용해서 신호로 처리한다.
단점은 만약에 TTTTTT와 같은 반복서열이 나올때, 9개와 10개를 구분하기 어렵다는 점이다.
만약 그렇게 인식이 되면 답안지가 밀려쓰게 되는거처럼 염기서열이 아예 바뀌어버리게 된다는 점이 있다.
장점은 4메가베이스페어정도를 한번이 읽을 수 있는 롱 리드를 지닌 점이다.
롱리드의 장점은 아래와 같다.
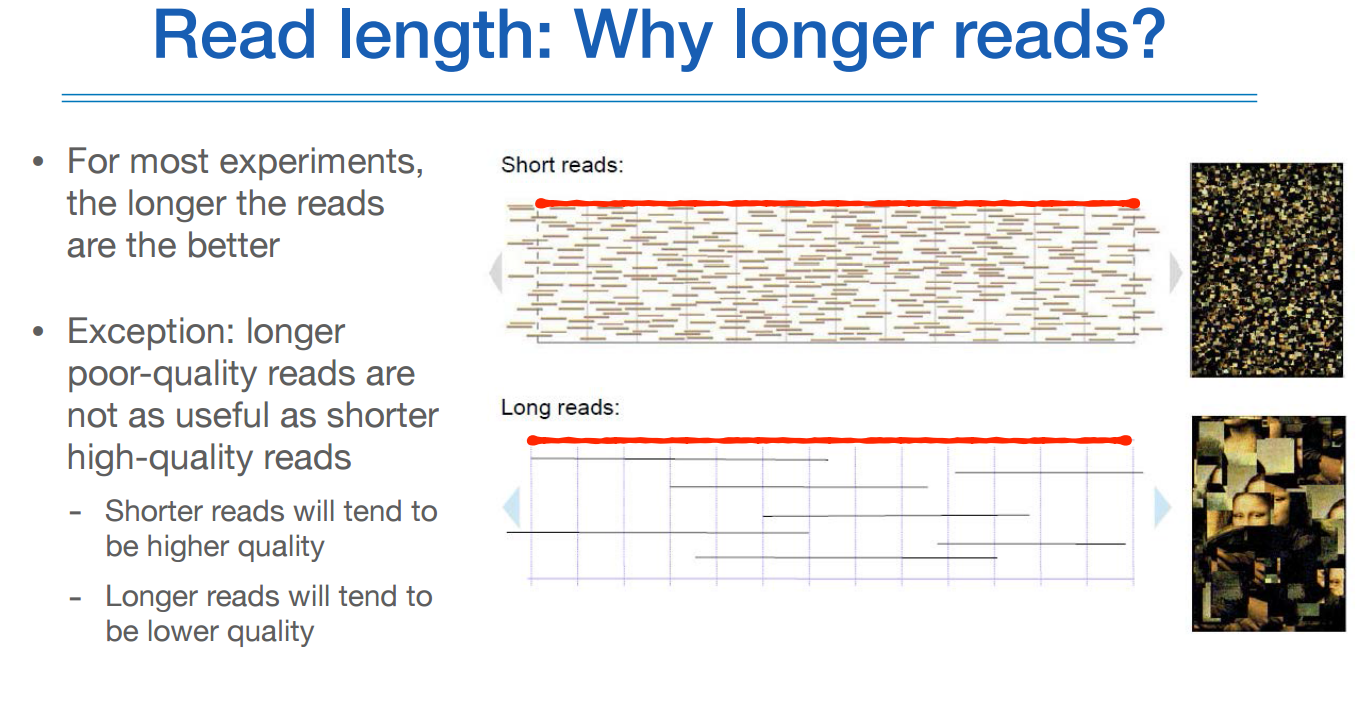
예를 들면 모나리자 그림의 퍼즐 조각을 맞출때, 1000피스짜리와 100피스짜리의 난이도를 완전히 다를것이다.
그럼에도 롱 리드는 그 자체가 정확하지 않아, 아직까지 전체적인 정확도가 높은 것은 숏리드이다.
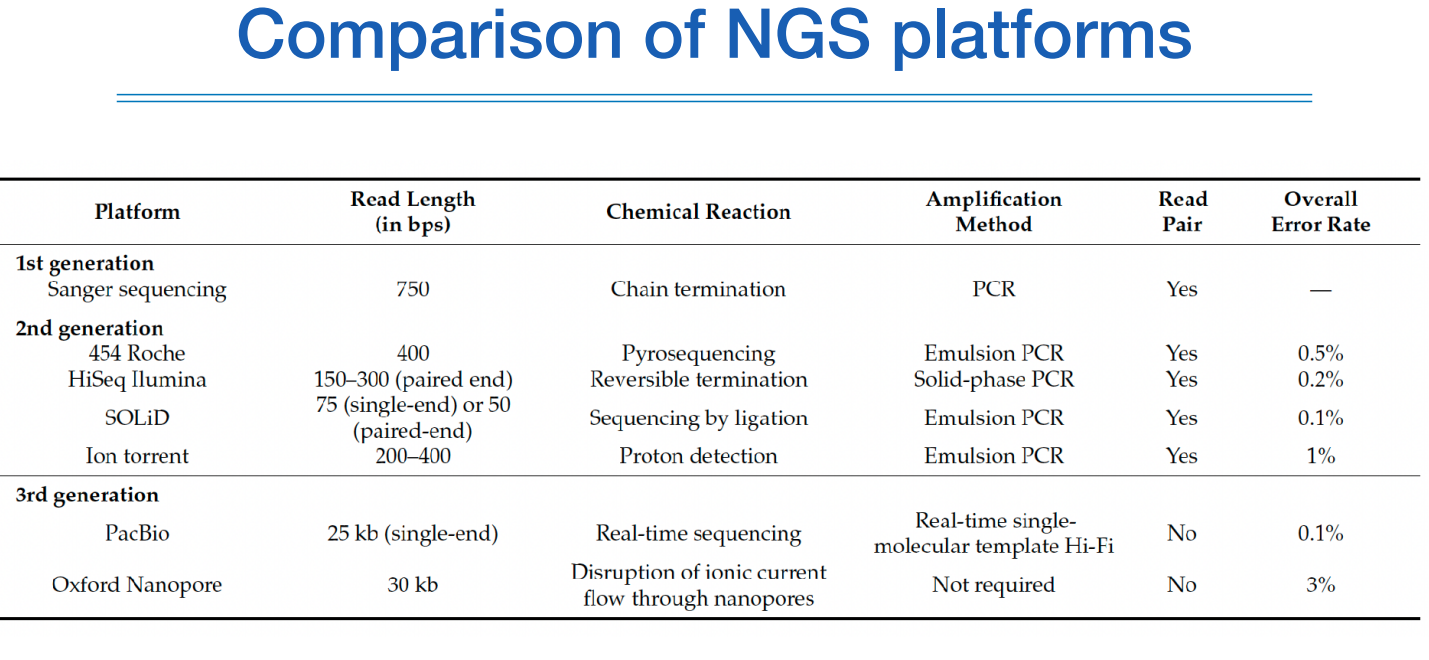
NGS 플랫폼의 비교를 한표로 보면 위와 같다.
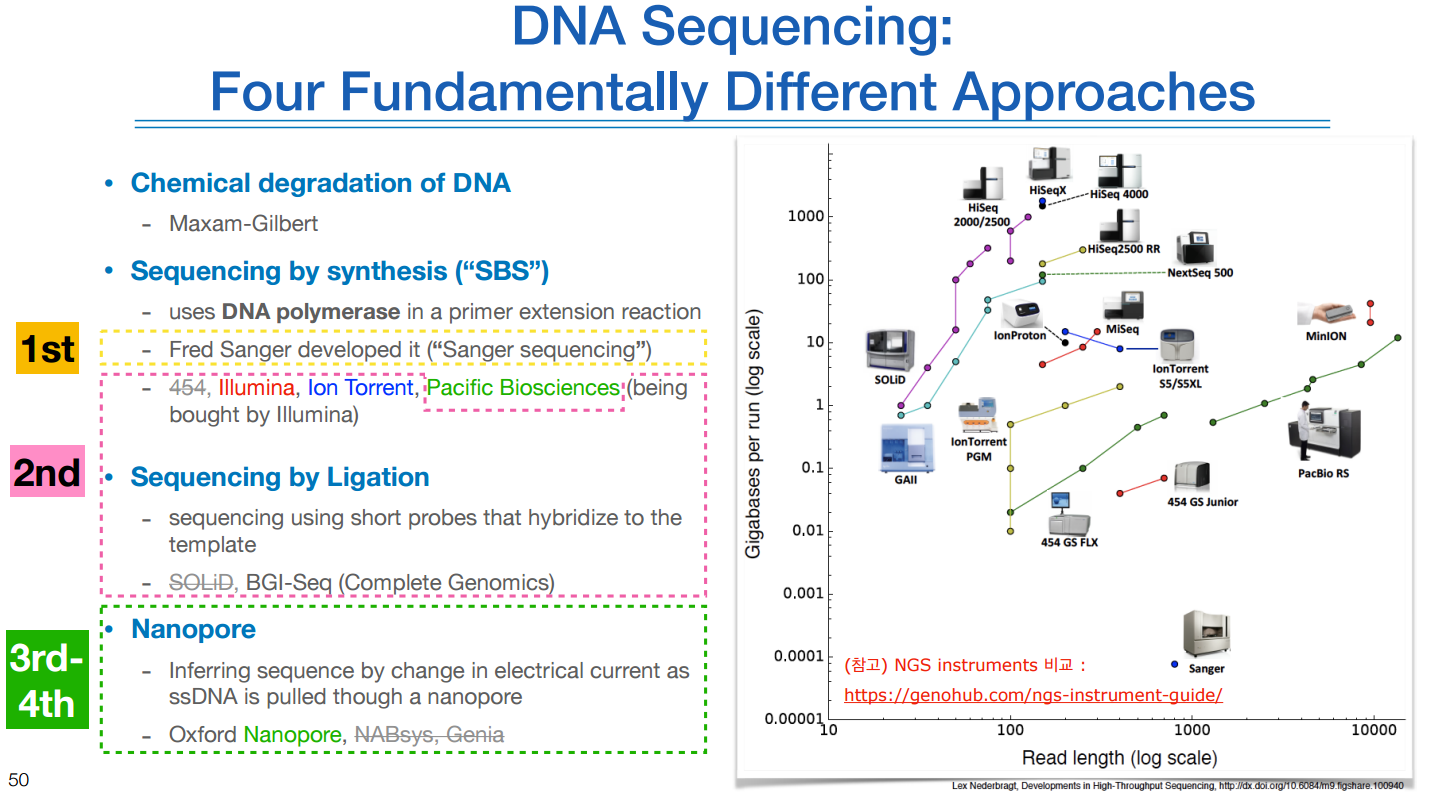
우측 그림을 보면 일루미나 시퀀서가 가장 높게 위치해있다.
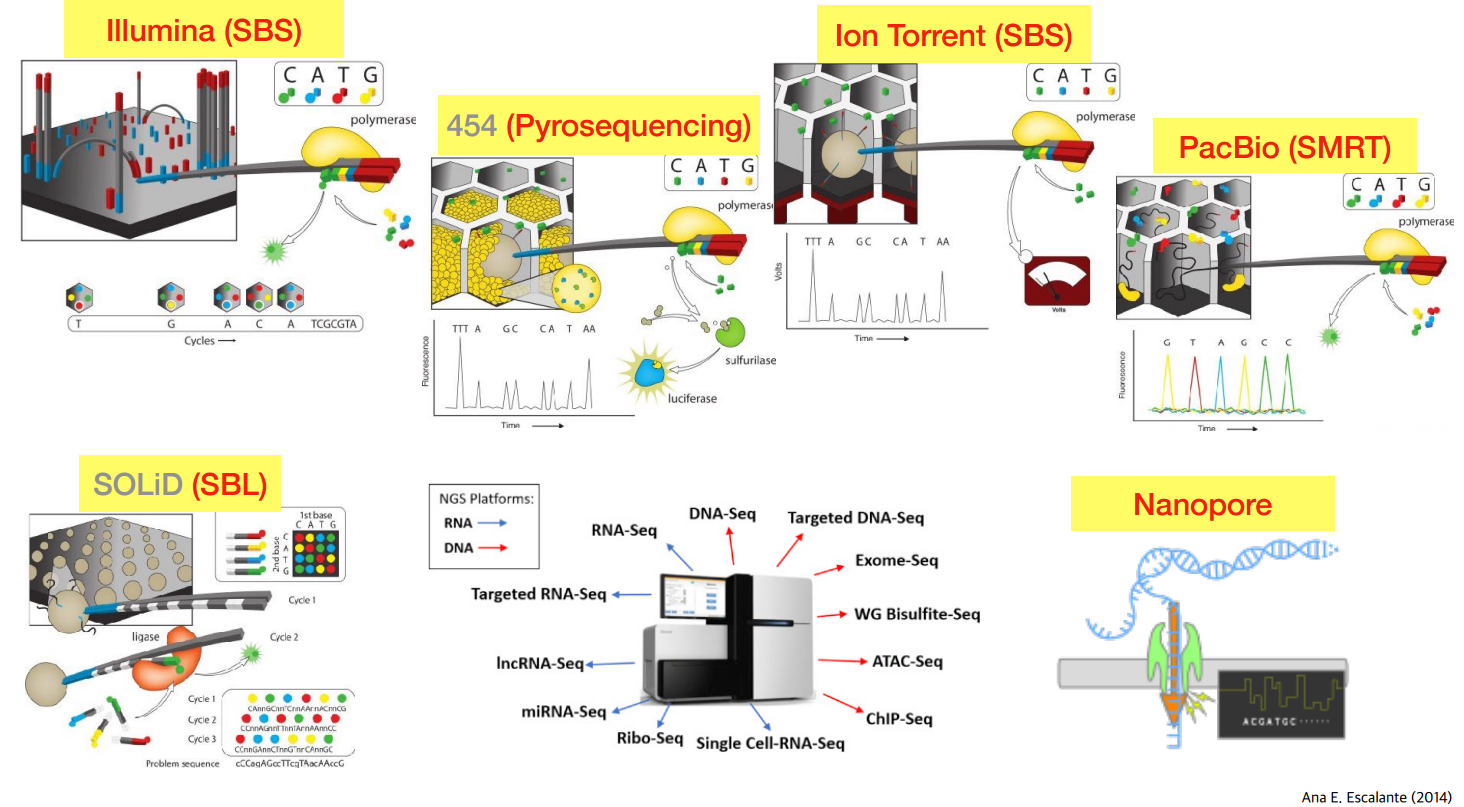
위 그림이 오늘 설명한 내용을 잘 담아놓은 그림이다.
끝!!
'medical' 카테고리의 다른 글
| [medical] 유전역학-개론 7주차, 유전자 변이에 대한 명명법 (0) | 2023.04.19 |
|---|---|
| [medical] 유전역학-개론 6주차, 차세대 시퀀싱(NGS) 데이터 분석 (1) | 2023.04.12 |
| [medical] 유전역학-개론 4주차, 전장유전체 연관분석(GWAS) (2) | 2023.03.28 |
| [medical] 유전역학-개론 3주차, 연구설계방법 (0) | 2023.03.22 |
| [medical] 유전역학-개론 2주차, 기초유전학 (0) | 2023.03.21 |



